


Help improve this workflow!
This workflow has been published but could be further improved with some additional meta data:- Keyword(s) in categories input, output, operation
You can help improve this workflow by suggesting the addition or removal of keywords, suggest changes and report issues, or request to become a maintainer of the Workflow .
#Snakemake workflow: ATAC-seq
This is using the standard Snakemake workflow template. Replace this text with a comprehensive description covering the purpose and domain. Insert your code into the respective folders, i.e.
scripts
,
rules
, and
envs
. Define the entry point of the workflow in the
Snakefile
and the main configuration in the
config.yaml
file.
This is the first version of ATAC-seq, using Bassing lab datasets dsb vs ctrl and no_DSB vs with_DSB
Authors
- chaodi([email protected])
Usage
Running on new respublica by: snakemake --latency-wait 10 -j 10 -p -c "sbatch --job-name={params.jobName} --mem={params.mem} -c {threads} --time=360 -e sbatch/{params.jobName}.e -o sbatch/{params.jobName}.o"
Workflow
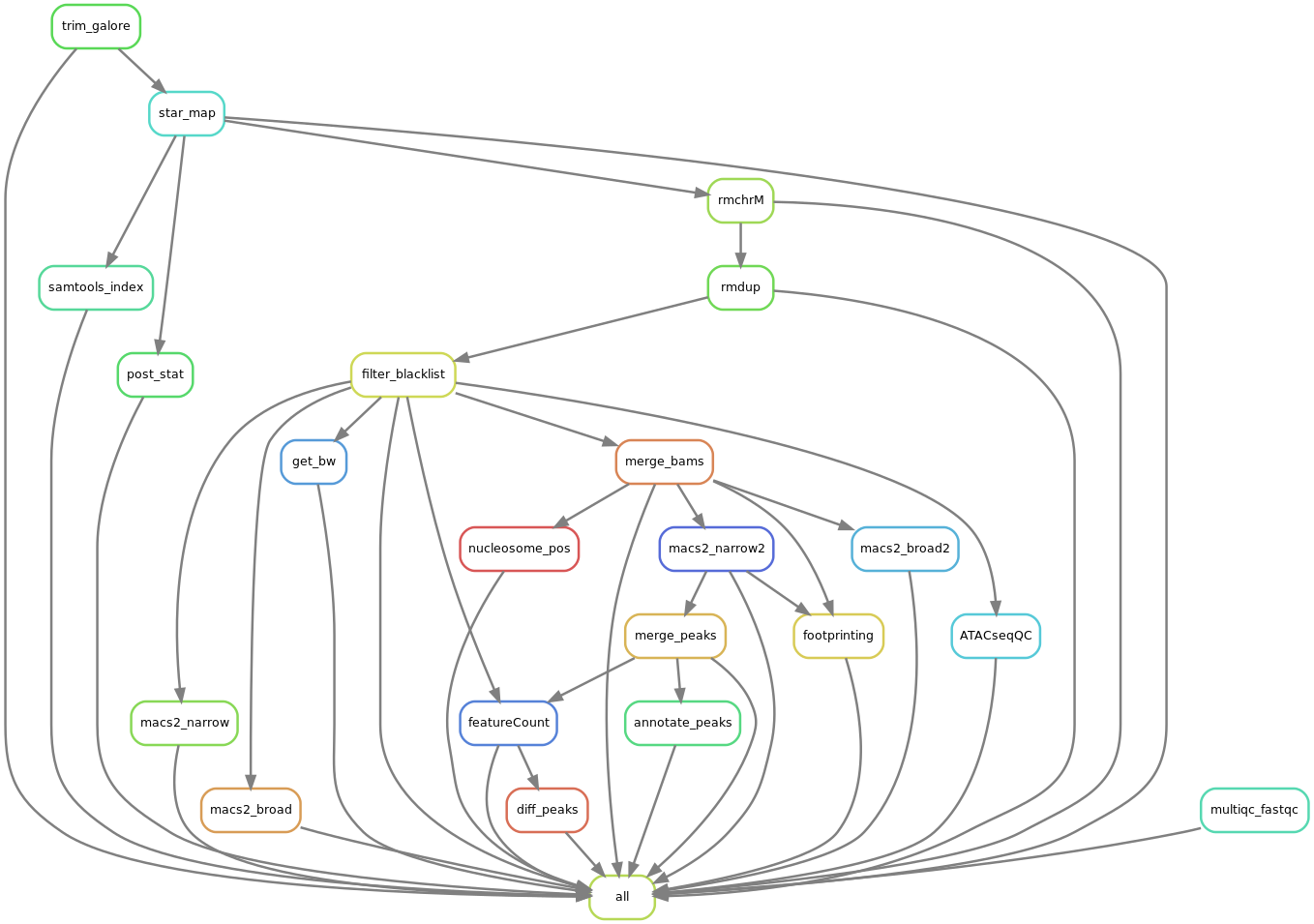
Code Snippets
13 14 15 16 | shell: ''' annotatePeaks.pl {input.merged_peaks} {params.genome} -annStats {output.anno_stats}> {output.annotated_peaks} 2>> {log} ''' |
11 12 | script: "../scripts/ATACseqQC.Rmd" |
12 13 | script: "../scripts/differential_peaks.Rmd" |
18 19 | script: "../scripts/featureCount_segments.R" |
12 13 14 15 16 | shell: ''' samtools view --threads {threads} -h {input.bam} | grep -v chrM | \ samtools sort --threads {threads} -O bam -o {output.rmChrM} &> {log} ''' |
28 29 30 31 32 | shell: ''' java -XX:ParallelGCThreads={threads} -jar ~/public/tools/picard-2.25.1-1/picard.jar MarkDuplicates --INPUT \ {input.rmChrM} --OUTPUT {output.rmdup} --METRICS_FILE {output.dups} --REMOVE_DUPLICATES true &> {log} ''' |
46 47 48 49 50 51 | shell: ''' bedtools intersect -v -abam {input.rmdup} -b {params.blacklist} > {output.rmBList} samtools index {output.rmBList} {output.index} ''' |
15 16 17 18 19 20 21 22 23 | shell: ''' # rgt-hint footprinting --atac-seq --paired-end --organism=mm10 \ --output-location=footprinting --output-prefix={wildcards.group} {input.merged_bam} {input.peak} &> {log} rgt-hint tracks --bc --bigWig --organism=mm10 --output-location=footprinting --output-prefix={wildcards.group} {input.merged_bam} {input.peak} &>> {log} rgt-motifanalysis matching --organism=mm10 --output-location=footprinting --input-files footprinting/{params.group_name}.bed &>> {log} ''' |
14 15 16 17 18 | shell: ''' # samtools merge --threads {threads} {output.merged_bam} {input.bam} &>> {log} samtools index {output.merged_bam} &>> {log} ''' |
14 15 16 17 18 | shell: ''' mergePeaks {input.allpeaks} > {output.merged_peaks} 2>> {log} sed 1d {output.merged_peaks} | awk 'BEGIN{{FS=OFS="\t"}} {{print $1"\t"$2"\t"$3"\t"$4"\t"$5}}' > {output.merged_peaks_saf} 2>> {log} ''' |
13 14 15 16 17 | shell: ''' java -Xmx95g -jar /home/dic/public/tools/hmmratac-1.2.10/share/hmmratac-1.2.10-1/HMMRATAC.jar -b {input.merged_bam} \ -i {input.bam_index} -g {params.genome_info} -o nucleosome_pos/"{wildcards.group}" --bedgraph TRUE &>{log} ''' |
12 13 14 15 16 | shell: ''' mkdir -p macs2_merged_bam/narrowPeak macs2 callpeak -f BAMPE -g mm --keep-dup all -n {wildcards.group} -t {input.merged_bam} --outdir {params.narrow_out} &> {log} ''' |
29 30 31 32 33 | shell: ''' mkdir -p macs2_merged_bam/broadPeak macs2 callpeak --broad -f BAMPE -g mm --keep-dup all -n {wildcards.group} -t {input.merged_bam} --outdir {params.broad_out} &>> {log} ''' |
12 13 14 15 16 | shell: ''' mkdir -p macs2/narrowPeak macs2 callpeak -B -f BAMPE -g mm --keep-dup all -n {wildcards.sample} -t {input.rmBList} --outdir {params.narrow_out} &> {log} ''' |
29 30 31 32 33 | shell: ''' mkdir -p macs2/broadPeak macs2 callpeak -B --broad -f BAMPE -g mm --keep-dup all -n {wildcards.sample} -t {input.rmBList} --outdir {params.broad_out} &>> {log} ''' |
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 | shell: ''' rm -f {output}; for i in {input}; do sampleName="$(basename $i .Log.final.out)"; cat $i | grep 'Number of input reads' | awk '{{print $6}}' > foo1; cat $i | grep 'Uniquely mapped reads number' | awk '{{print $6}}' > foo2; cat $i | grep 'Number of reads mapped to multiple loci' | awk '{{print $9}}' > foo3; cat $i | grep "Uniquely mapped reads %" | awk '{{print $6}}' > foo4; cat $i | grep "% of reads mapped to multiple loci"|awk '{{print $9}}' > foo5; paste foo1 foo2 foo3 foo4 foo5 | awk '{{print "'$sampleName'\t"$1"\t"$2+$3"\t"$4+$5}}' >> {output.mappedReads} done cat {output.mappedReads} | awk '{{print $1"\t"1000000/$2}}' > {output.rpmFactor} sed -i '1isample\ttotal_reads\tmapped_reads\t%mapped' {output.mappedReads}; rm -f foo* ''' |
15 16 17 18 19 20 21 | shell: "STAR --runThreadN {threads} --genomeDir {params.genome_dir} " "--outFileNamePrefix STAR_align/{wildcards.sample}. --outSAMtype BAM SortedByCoordinate " "--outSAMmapqUnique 255 --outFilterMultimapNmax 1 " "--quantMode GeneCounts --sjdbGTFfile {params.annotation} " "--readFilesIn {input} --outSJfilterReads Unique --readFilesCommand gunzip -c && " "mv STAR_align/{wildcards.sample}.Aligned.sortedByCoord.out.bam STAR_align/{wildcards.sample}.bam" |
31 32 | shell: "samtools index {input} {output}" |
15 16 17 18 19 20 | shell: ''' trim_galore --cores {threads} --gzip --fastqc --paired -o {params.outdir} {input} &> {log} \ mv trimmed_fq/{wildcards.sample}_1_val_1.fq.gz {output.fq1} &>> {log} \ mv trimmed_fq/{wildcards.sample}_2_val_2.fq.gz {output.fq2} &>> {log} ''' |
31 32 33 34 35 | shell: ''' rm -r {params.outdir} multiqc trimmed_fq/ -o {params.outdir} &> {log} ''' |
15 16 | # replace with path where you want the results be knitr::opts_knit$set(root.dir="/home/dic/bassing_lab/users/dic/ATAC-seq/workflow/") |
20 21 22 23 24 25 26 27 28 29 30 31 32 33 | # log <- file(snakemake@log[[1]], open="wt") # sink(log) # sink(log, type="message") setwd("/home/dic/bassing_lab/users/dic/ATAC-seq/workflow/") library(ATACseqQC) library(BSgenome.Mmusculus.UCSC.mm10) library(TxDb.Mmusculus.UCSC.mm10.knownGene) # library(phstCons60way.UCSC.mm10) # library(BSgenome.Hsapiens.UCSC.hg19) # library(TxDb.Hsapiens.UCSC.hg19.knownGene) # library(phastCons100way.UCSC.hg19) library(Rsamtools) library(ChIPpeakAnno) library(MotifDb) |
38 39 40 41 42 | # bamfile <- system.file("extdata", "GL1.bam", package="ATACseqQC", mustWork=TRUE) # bamfile.labels <- gsub(".bam", "", basename(bamfile)) bamfile <- snakemake@input[["rmBList"]] bamfile.labels <- sub(".rmBList.bam", "", basename(bamfile)) |
47 | estimateLibComplexity(readsDupFreq(bamfile)) |
53 | fragSizeDist(bamfile, bamfile.labels) |
64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 | ## bamfile tags to be read in possibleTag <- list("integer"=c("AM", "AS", "CM", "CP", "FI", "H0", "H1", "H2", "HI", "IH", "MQ", "NH", "NM", "OP", "PQ", "SM", "TC", "UQ"), "character"=c("BC", "BQ", "BZ", "CB", "CC", "CO", "CQ", "CR", "CS", "CT", "CY", "E2", "FS", "LB", "MC", "MD", "MI", "OA", "OC", "OQ", "OX", "PG", "PT", "PU", "Q2", "QT", "QX", "R2", "RG", "RX", "SA", "TS", "U2")) bamTop100 <- scanBam(BamFile(bamfile, yieldSize = 100), param = ScanBamParam(tag=unlist(possibleTag)))[[1]]$tag tags <- names(bamTop100)[lengths(bamTop100)>0] tags ## files will be output into outPath outPath <- paste0("STAR_align_filter/splited/", bamfile.labels) dir.create(outPath) ## shift the coordinates of 5'ends of alignments in the bam file seqlev <- "chr1" ## subsample data for quick run seqinformation <- seqinfo(TxDb.Mmusculus.UCSC.mm10.knownGene) which <- as(seqinformation[seqlev], "GRanges") gal <- readBamFile(bamfile, tag=tags, which=which, asMates=TRUE) shiftedBamfile <- file.path(outPath, "shifted.bam") gal1 <- shiftGAlignmentsList(gal, outbam=shiftedBamfile) |
96 97 98 99 100 101 | txs <- transcripts(TxDb.Mmusculus.UCSC.mm10.knownGene) tsse <- TSSEscore(gal1, txs) tsse$TSSEscore plot(100*(-9:10-.5), tsse$values, type="b", xlab="distance to TSS", ylab="aggregate TSS score") |
109 110 111 112 113 114 115 116 117 | ## run program for chromosome 1 only txs <- txs[seqnames(txs) %in% "chr1"] genome <- Mmusculus ## split the reads into NucleosomeFree, mononucleosome, ## dinucleosome and trinucleosome. ## and save the binned alignments into bam files. objs <- splitGAlignmentsByCut(gal1, txs=txs, genome=genome, outPath = outPath) ## list the files generated by splitGAlignmentsByCut. dir(outPath) |
124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 | bamfiles <- file.path(outPath, c("NucleosomeFree.bam", "mononucleosome.bam", "dinucleosome.bam", "trinucleosome.bam")) ## Plot the cumulative percentage of tag allocation in nucleosome-free ## and mononucleosome bam files. cumulativePercentage(bamfiles[1:2], as(seqinformation["chr1"], "GRanges")) TSS <- promoters(txs, upstream=0, downstream=1) TSS <- unique(TSS) ## estimate the library size for normalization (librarySize <- estLibSize(bamfiles)) ## calculate the signals around TSSs. NTILE <- 101 dws <- ups <- 1010 sigs <- enrichedFragments(gal=objs[c("NucleosomeFree", "mononucleosome", "dinucleosome", "trinucleosome")], TSS=TSS, librarySize=librarySize, seqlev=seqlev, TSS.filter=0.5, n.tile = NTILE, upstream = ups, downstream = dws) ## log2 transformed signals sigs.log2 <- lapply(sigs, function(.ele) log2(.ele+1)) #plot heatmap featureAlignedHeatmap(sigs.log2, reCenterPeaks(TSS, width=ups+dws), zeroAt=.5, n.tile=NTILE) ## get signals normalized for nucleosome-free and nucleosome-bound regions. out <- featureAlignedDistribution(sigs, reCenterPeaks(TSS, width=ups+dws), zeroAt=.5, n.tile=NTILE, type="l", ylab="Averaged coverage") ## rescale the nucleosome-free and nucleosome signals to 0~1 range01 <- function(x){(x-min(x))/(max(x)-min(x))} out <- apply(out, 2, range01) matplot(out, type="l", xaxt="n", xlab="Position (bp)", ylab="Fraction of signal") axis(1, at=seq(0, 100, by=10)+1, labels=c("-1K", seq(-800, 800, by=200), "1K"), las=2) abline(v=seq(0, 100, by=10)+1, lty=2, col="gray") |
180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 | ## foot prints CTCF <- query(MotifDb, c("CTCF")) CTCF <- as.list(CTCF) print(CTCF[[1]], digits=2) sigs <- factorFootprints(shiftedBamfile, pfm=CTCF[[1]], genome=genome, ## Don't have a genome? ask ?factorFootprints for help min.score="90%", seqlev=seqlev, upstream=100, downstream=100) featureAlignedHeatmap(sigs$signal, feature.gr=reCenterPeaks(sigs$bindingSites, width=200+width(sigs$bindingSites[1])), annoMcols="score", sortBy="score", n.tile=ncol(sigs$signal[[1]])) |
17 18 | # replace with path where you want the results be knitr::opts_knit$set(root.dir="/home/dic/bassing_lab/users/dic/ATAC-seq/workflow") |
22 23 24 25 26 | # set home dir when test in console library(DESeq2) library(ROTS) library(ggplot2) library(dplyr) |
35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 | cts <- read.table(snakemake@input[["featureCount"]], header=TRUE, row.names=1, check.names=FALSE) colnames(cts) <- sub(".rmBList.bam","",colnames(cts)) coldata <- read.table(snakemake@input[["sample_table"]], header=TRUE, row.names="sample",sep="\t", check.names=FALSE) coldata <- coldata[,c(3:4)] head(coldata) # load the counts with no design dds <- DESeqDataSetFromMatrix(countData=cts, colData=coldata, design=~group) # genes have at least 10 reads in at least 2 samples dds <- dds[rowSums(counts(dds) >= 10) >= 2,] # normalization and pre-processing dds <- DESeq(dds) # raw count normalization norm_counts <- counts(dds, normalized=TRUE) # count transformation, log2 scale, either rlog or vst vsd <- vst(dds, blind=FALSE) vsd_data <- as.data.frame(assay(vsd)) cat("The data values after transformation:\n") head(vsd_data) |
63 64 65 66 67 68 69 | pcaData <- plotPCA(vsd, intgroup=c("group"), returnData=TRUE) percentVar <- round(100 * attr(pcaData, "percentVar")) ggplot(pcaData, aes(PC1, PC2, color=group)) + geom_point(size =3)+ xlab(paste0("PC1: ", percentVar[1], "% variance")) + ylab(paste0("PC2: ", percentVar[2], "% variance")) + coord_fixed() |
77 78 79 80 | resultsNames(dds) res <- results (dds) table(res$padj<0.1) res[!is.na(res$padj) & res$padj<0.1,] |
90 91 92 93 94 95 96 97 | groups = c(rep(0,2), rep(1,2)) rots.out = ROTS(data = vsd_data, groups = groups, B = 1000, seed = 1234) summary(rots.out, fdr = 0.05) cat("Number of differential peaks with FDR<0.1:") length(rots.out$FDR[rots.out$FDR<0.1]) plot(rots.out, fdr = 0.1, type = "volcano") cat("Reproducibility Z-scores below 2 indicate that the data or the statistics are not sufficient for reliable detection!!!") rots.out$Z |
2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 | library(Rsubread) library(dplyr) library(mgsub) log <- file(snakemake@log[[1]], open="wt") sink(log) sink(log, type="message") ## run all bams together bamfiles <- snakemake@input[["bamfiles"]] # bamfiles <- paste0("./STAR_align_filter/", as.vector(samples$sample),".rmBList.bam") ## run one bam file # bamfiles <- snakemake@input[["bamfile"]] RPFcounts <- featureCounts(files=bamfiles, annot.ext=snakemake@input[['merged_peaks_saf']], annot.inbuilt = "mm10", isGTFAnnotationFile=FALSE, minOverlap = 1, fracOverlap=snakemake@params[["fracOverlap"]], fracOverlapFeature=snakemake@params[["fracOverlapFeature"]], allowMultiOverlap=TRUE, strandSpecific=snakemake@params[["strand"]], countMultiMappingReads=FALSE, juncCounts=TRUE, isPairedEnd=TRUE, nthreads=snakemake@threads[[1]]) write.table(RPFcounts$counts, file=snakemake@output[["featureCount"]], sep="\t", quote=F, row.names = TRUE, col.names = NA) |
Support
Do you know this workflow well? If so, you can
request seller status , and start supporting this workflow.
- Share:
-

-

-

Created: 1yr ago
Updated: 1yr ago
Maitainers:
public
URL:
https://github.com/chaodi51/ATAC-seq
Name:
atac-seq
Version:
2
Other Versions:
Downloaded:
0
Copyright:
Public Domain
License:
MIT License
Keywords:
BSgenome.Hsapiens.UCSC.hg19
BSgenome.Mmusculus.UCSC.mm10
TxDb.Hsapiens.UCSC.hg19.knownGene
TxDb.Mmusculus.UCSC.mm10.knownGene
macs2
PCAtools
ROTS
Rsamtools
ATACseqQC
BEDTools
ChIPpeakAnno
DESeq2
FeatureCounts
homer
MotifDb
MultiQC
Picard
Rsubread
SAMtools
Snakemake
STAR
dplyr
ggplot2
mgsub
Trim_Galore
ATAC-seq
- Future updates
Related Workflows

ENCODE pipeline for histone marks developed for the psychENCODE project
psychip pipeline is an improved version of the ENCODE pipeline for histone marks developed for the psychENCODE project.
The o...

Near-real time tracking of SARS-CoV-2 in Connecticut
Repository containing scripts to perform near-real time tracking of SARS-CoV-2 in Connecticut using genomic data. This pipeli...

snakemake workflow to run cellranger on a given bucket using gke.
A Snakemake workflow for running cellranger on a given bucket using Google Kubernetes Engine. The usage of this workflow ...

ATLAS - Three commands to start analyzing your metagenome data
Metagenome-atlas is a easy-to-use metagenomic pipeline based on snakemake. It handles all steps from QC, Assembly, Binning, t...
raw sequence reads
Genome assembly
Annotation track
checkm2
gunc
prodigal
snakemake-wrapper-utils
MEGAHIT
Atlas
BBMap
Biopython
BioRuby
Bwa-mem2
cd-hit
CheckM
DAS
Diamond
eggNOG-mapper v2
MetaBAT 2
Minimap2
MMseqs
MultiQC
Pandas
Picard
pyfastx
SAMtools
SemiBin
Snakemake
SPAdes
SqueezeMeta
TADpole
VAMB
CONCOCT
ete3
gtdbtk
h5py
networkx
numpy
plotly
psutil
utils
metagenomics

RNA-seq workflow using STAR and DESeq2
This workflow performs a differential gene expression analysis with STAR and Deseq2. The usage of this workflow is described ...

This Snakemake pipeline implements the GATK best-practices workflow
This Snakemake pipeline implements the GATK best-practices workflow for calling small germline variants. The usage of thi...