









bigr_epicure_pipeline
Snakemake pipeline for Epicure analyses: Chip-Seq, Atac-Seq, Cut&Tag, Cut&Run, MeDIP-Seq, 8-OxoG-Seq
Summary
Usage
Installation (following Snakemake-Workflows guidelines)
note: This action has already been done for you if you work at Gustave Roussy. See at the end of this section
- Install snakemake and snakedeploy with mamba package manager. Given that Mamba is installed, run:
mamba create -c conda-forge -c bioconda --name bigr_epicure_pipeline snakemake snakedeploy pandas
- Ensure your conda environment is activated in your bash terminal:
conda shell.bash activate bigr_epicure_pipeline
Alternatively, if you work at Gustave Roussy, you can use our shared environment:
conda shell.bash activate /mnt/beegfs/pipelines/unofficial-snakemake-wrappers/shared_conda/bigr_epicure_pipeline
Deployment (following Snakemake-Workflows guidelines)
note: This action has been made easier for you if you work at Gustave Roussy. See at the end of this section
Given that Snakemake and Snakedeploy are installed and available (see Installation ), the workflow can be deployed as follows.
- Go to your working directory:
cd path/to/my/project
- Deploy workflow:
snakedeploy deploy-workflow https://github.com/tdayris/bigr_epicure_pipeline . --tag v0.4.0
Snakedeploy will create two folders
workflow
and
config
. The former contains the deployment of the chosen workflow as a
Snakemake module
, the latter contains configuration files which will be modified in the next step in order to configure the workflow to your needs. Later, when executing the workflow, Snakemake will automatically find the main
Snakefile
in the
workflow
subfolder.
- Consider to put the exact version of the pipeline and all modifications you might want perform under version control. e.g. by managing it via a (private) Github repository
Configure workflow (following Snakemake-Workflows guidelines)
See dedicated
config/README.md
file for dedicated explanations of all options and consequences.
Run workflow (following Snakemake-Workflows guidelines)
note: This action has been made easier for you if you work at Gustave Roussy. See at the end of this section
Given that the workflow has been properly deployed and configured, it can be executed as follows.
Fow running the workflow while deploying any necessary software via conda (using the Mamba package manager by default), run Snakemake with
snakemake --cores all --use-conda
Alternatively, for users at Gustave Roussy, you may use:
bash workflow/scripts/misc/bigr_launcher.sh
Snakemake will automatically detect the main
Snakefile
in the
workflow
subfolder and execute the workflow module that has been defined by the deployment in step 2.
For further options, e.g. for cluster and cloud execution, see the docs .
Generate report (following Snakemake-Workflows guidelines)
After finalizing your data analysis, you can automatically generate an interactive visual HTML report for inspection of results together with parameters and code inside of the browser via
snakemake --report report.zip
The resulting
report.zip
file can be passed on to collaborators, provided as a supplementary file in publications, or uploaded to a service like
Zenodo
in order to obtain a citable
DOI
.
Open-on-demand Flamingo
This section describes the pipeline usage through Open-on-demand web-interface at Flamingo, Gustave Roussy.
Open-on-demand job composer
Log-in to Flamingo Open-On-Demand web-page using your institutional user name and password. On the main page, click on "Job", then, in the drop-down menu, hit "Job Composer"

Create a new template
On the top of the new page, in the grey bar menu, hit "Template". Then use the light blue button "+ New Template".

An empty template for appears:

Fill the "Path" section with the following path:
/mnt/beegfs/pipelines/bigr_epicure_pipeline/open-on-demand-templates
. It is very important that you do not change this path. Fill the rest of the template as you like.

Hit the white "save" button at the bottom left of the form.
Edit the template with your current work
Back to the "Template" page we saw earlier, a new line should be displayed with our new template. Click on the line and the right section of the page should display dedicated informations about this template.

Name it as you like, but do not create a job now! Scroll down and hit the white button: "Open Dir"

Edit
bigr_launcher.sh
Click on the file
bigr_launcher.sh
and hit the white button: "Edit".

Follow comments in grey and edit the line 11 to 30 as you'd like. You can refer to this page to know the latest version of this pipeline. You should always be using the latest version of this pipeline when starting a new project.

Launch a new job
Back to the Job Composer window, you can now click "+ New Job", "From Template" and select the template we just made.

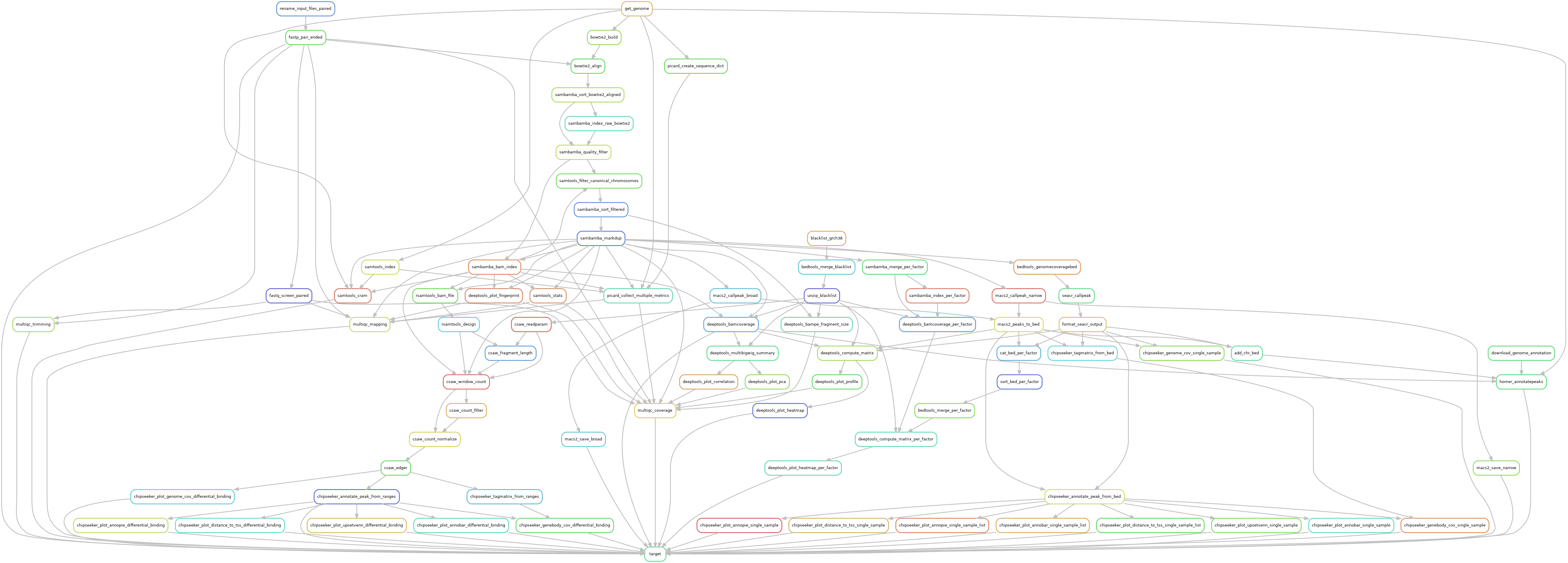
Pipeline description
note: The following steps may not be perform in that exact order.
Pre-pocessing
| Step | Tool | Documentation | Reason |
|---|---|---|---|
| Download genome sequence | curl | Snakemake-Wrapper: download-sequence | Ensure genome sequence are consistent in Epicure analyses |
| Download genome annotation | curl | Snakemake-Wrapper: download-annotation | Ensure genome annotation are consistent in Epicure analyses |
| Download blacklised regions | wget | Ensure blacklist regions are consistent in Epicure analyses | |
| Trimming + QC | Fastp | Snakemake-Wrapper: fastp | Perform read quality check and corrections, UMI, adapter removal, QC before and after trimming |
| Quality Control | FastqScreen | Snakemake-Wrapper: fastq-screen | Perform library quality check |
| Download fastq screen indexes | wget | Ensure fastq_screen reports are the same among Epicure analyses |
Read mapping
| Step | Tool | Documentation | Reason |
|---|---|---|---|
| Indexation | Bowtie2 | Snakemake-Wrapper: bowtie2-build | Index genome for up-coming read mapping |
| Mapping | Bowtie2 | Snakemake-Wrapper: bowtie2-align | Align sequenced reads on the genome |
| Filtering | Sambamba | Snakemake-Wrapper: sambamba-sort | Sort alignment over chromosome position, this reduce up-coming required computing resources, and reduce alignment-file size. |
| Filtering | Sambamba | Snakemake-Wrapper: sambamba-view | Remove non-canonical chromosomes and mapping belonging to mitochondrial chromosome. Remove low quality mapping. |
| Filtering | Sambamba | Snakemake-Wrapper: sambamba-markdup | Remove sequencing duplicates. |
| Filtering | DeepTools | Snakemake-Wrapper: sambamba-sort | For Atac-seq only. Reads on the positive strand should be shifted 4 bp to the right and reads on the negative strand should be shifted 5 bp to the left as in Buenrostro et al. 2013 . |
| Archive | Sambamba | Snakemake-Wrapper: sambamba-view | Compress alignment fil in CRAM format in order to reduce archive size. |
| Quality Control | Picard | Snakemake-Wrapper: picard-collect-multiple-metrics | Summarize alignments, GC bias, insert size metrics, and quality score distribution. |
| Quality Control | Samtools | Snakemake-Wrapper: samtools-stats | Summarize alignment metrics. Performed before and after mapping-post-processing in order to highlight possible bias. |
| Quality Control | DeepTools | Snakemake-Wrapper: deeptools-fingerprint | Control imuno precipitation signal specificity. |
| GC-bias correction | DeepTools | Official Documentation | Filter regions with abnormal GC-Bias as decribed in Benjamini & Speed, 2012 . OxiDIP-Seq only. |
Coverage
| Step | Tool | Documentation | Reason |
|---|---|---|---|
| Coverage | DeepTools | Snakemake-Wrapper: deeptools-bamcoverage | Compute genome coverage, normalized to 1M reads |
| Coverage | MEDIPS | Official documentation | Compute genome coverage with CpG density correction using MEDIPS (MeDIP-Seq only) |
| Scaled-Coverage | DeepTools | Snakemake-Wrapper: deeptools-computematrix | Calculate scores per genomic regions. Used for heatmaps and profile coverage plots. |
| Depth | DeepTools | Snakemake-Wrapper: deeptools-plotheatmap | Assess the sequencing depth of given samples |
| Coverage | CSAW | Official documentation | Count and filter reads over sliding windows. |
| Filter | CSAW | Official documentation | Filter reads over background signal. |
| Quality Control | DeepTools | Snakemake-Wrapper: deeptools-plotprofile | Plot profile scores over genomic regions |
| Quality Control | DeepTools | Official Documentation | Plot principal component analysis (PCA) over bigwig coverage to assess sample dissimilarity. |
| Quality Control | DeepTools | Official Documentation | Plot sample correlation based on the coverage analysis. |
| Coverage | BedTools | Snakemake-Wrapper: bedtools-genomecov | Estimate raw coverage over the genome |
Peak-Calling
| Step | Tool | Documentation | Reason |
|---|---|---|---|
| Peak-Calling | Mac2 | Snakemake-Wrapper: macs2-callpeak | Search for significant peaks |
| Heatmap | DeepTools | Snakemake-Wrapper: deeptools-plotheatmap | Plot heatmap and peak coverage among samples |
| FRiP score | Manually assessed | Compute Fragment of Reads in Peaks to assess signal/noise ratio. | |
| Peak-Calling | SEACR | Official Documentation | Search for significant peaks in Cut&Tag or Cut&Run |
Differential Peak Calling
| Step | Tool | Documentation | Reason |
|---|---|---|---|
| Peak-Calling | MEDIPS | Official documentation | Search for significant variation in peak coverage with EdgeR (MeDIP-Seq only) |
| Normalization | CSAW | Official documentation | Correct composition bias and variable library sizes. |
| Differential Binding | CSAW-EdgeR | Official documentation | Call for differentially covered windows |
Peak annotation
| Step | Tool | Documentation | Reason |
|---|---|---|---|
| Annotation | MEDIPS | Official Documentation | Annotate peaks with Ensembl-mart through MEDIPS (MeDIP-Seq only) |
| Annotation | Homer | Snakemake-Wrapper: homer-annotatepeaks | Performing peak annotation to associate peaks with nearby genes. |
| Annotation | CHiP seeker | Official Documentation | Performing peak annotation to associate peaks with nearby genes. |
| Quality Control | CHiP Seeker | Official Documentation | Perform region-wise barplot graph. |
| Quality Control | CHiP Seeker | Official Documentation | Perform region-wise upset-plot. |
| Quality Control | CHiP Seeker | Official Documentation | Visualize distribution of TF-binding loci relative to TSS |
Motifs
| Step | Tool | Documentation | Reason |
|---|---|---|---|
| De-novo | Homer | Official Documentation | Perform de novo motifs discovery. |
Material and Methods
ChIP-Seq

Reads are trimmed using
fastp
version
0.23.2
, using default parameters. Trimmed reads are mapped over the genome of interest defined in the configuration file, using
bowtie2
version
2.5.1
, using
--very-sensitive
parameter to increase mapping specificity.
Mapped reads with a quality lower than 30 are dropped out using
Sambamba
version
1.0
with parameter
--filter 'mapping_quality >= 30'
. In case of pair-ended library, orphan reads a dropped out using
--filter 'not (unmapped or mate_is_unmapped)'
in
Sambamba
version
1.0
. Remaining reads are filtered over canonical chromosomes, using
Samtools
version
1.17
. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome,
BedTools
version
2.31.0
with
-wa -sorted
as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were filtered out, using
Sambamba
version
1.0
using the optional parameter
--remove-duplicates --overflow-list-size 600000
.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0 , and Samtools version 1.17 .
Genome coverage was assessed with
DeepTools
version
3.5.2
. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. using the following optional parameters
--normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY
. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by
Macs2
version
2.2.7.1
using both broad and narrow options. Pair-ended libraries recieved the
--format BAMPE
parameters, while single-ended libraries used estimated fragment size provided in input.
The peak annotation was done using ChIPSeeker version 1.34.0 , using Org.eg.db annotations version 3.16.0 .
De novo
motif discovery was made with
Homer
version
4.11
over called peaks, using the following optional parameters
-size 200 -homer2
over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed using
CSAW
version
1.32.0
. Input signal (if available) was used to find signal of interest over background noise. Or else, a large binning was defined over mappable genome regions to define a background noise track to use as an input signal. In any case, a log2 of 1.1 between the background and the sample of interest was used as a threshold value to identify regions of interest. Data were normalized over efficiency biases as described in
CSAW documentation
, since libraries of the same size can still have composition bias. Differential binded sites were called using EdgeR over the statistical formulas described in the configuration file. The
prior N
was defined over 20 degrees of freedom in the GLM test. FDR values from EdgeR were corrected using a region merge accross gene annotations to account for related binding sites. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3 . Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.0.0 .
MNase-seq
Reads are trimmed using
fastp
version
0.23.2
, using default parameters. Trimmed reads are mapped over the genome of interest defined in the configuration file, using
bowtie2
version
2.5.1
, using
--very-sensitive
parameter to increase mapping specificity.
Mapped reads with a quality lower than 30 are dropped out using
Sambamba
version
1.0
with parameter
--filter 'mapping_quality >= 30'
. In case of pair-ended library, orphan reads a dropped out using
--filter 'not (unmapped or mate_is_unmapped)'
in
Sambamba
version
1.0
. Remaining reads are filtered over canonical chromosomes, using
Samtools
version
1.17
. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome,
BedTools
version
2.31.0
with
-wa -sorted
as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were filtered out, using
Sambamba
version
1.0
using the optional parameter
--remove-duplicates --overflow-list-size 600000
.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0 , and Samtools version 1.17 .
Genome coverage was assessed with
DeepTools
version
3.5.2
. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. MNase specificity was also taken into account using the following optional parameters
--normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY --MNase
. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by
Macs2
version
2.2.7.1
using both broad and narrow options. Pair-ended libraries recieved the
--format BAMPE
parameters, while single-ended libraries used estimated fragment size provided in input.
The peak annotation was done using ChIPSeeker version 1.34.0 , using Org.eg.db annotations version 3.16.0 .
De novo
motif discovery was made with
Homer
version
4.11
over called peaks, using the following optional parameters
-size 200 -homer2
over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed using
CSAW
version
1.32.0
. Input signal (if available) was used to find signal of interest over background noise. Or else, a large binning was defined over mappable genome regions to define a background noise track to use as an input signal. In any case, a log2 of 1.1 between the background and the sample of interest was used as a threshold value to identify regions of interest. Data were normalized over efficiency biases as described in
CSAW documentation
, since libraries of the same size can still have composition bias. Differential binded sites were called using EdgeR over the statistical formulas described in the configuration file. The
prior N
was defined over 20 degrees of freedom in the GLM test. FDR values from EdgeR were corrected using a region merge accross gene annotations to account for related binding sites. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3 . Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.0.0 .
Gro-seq
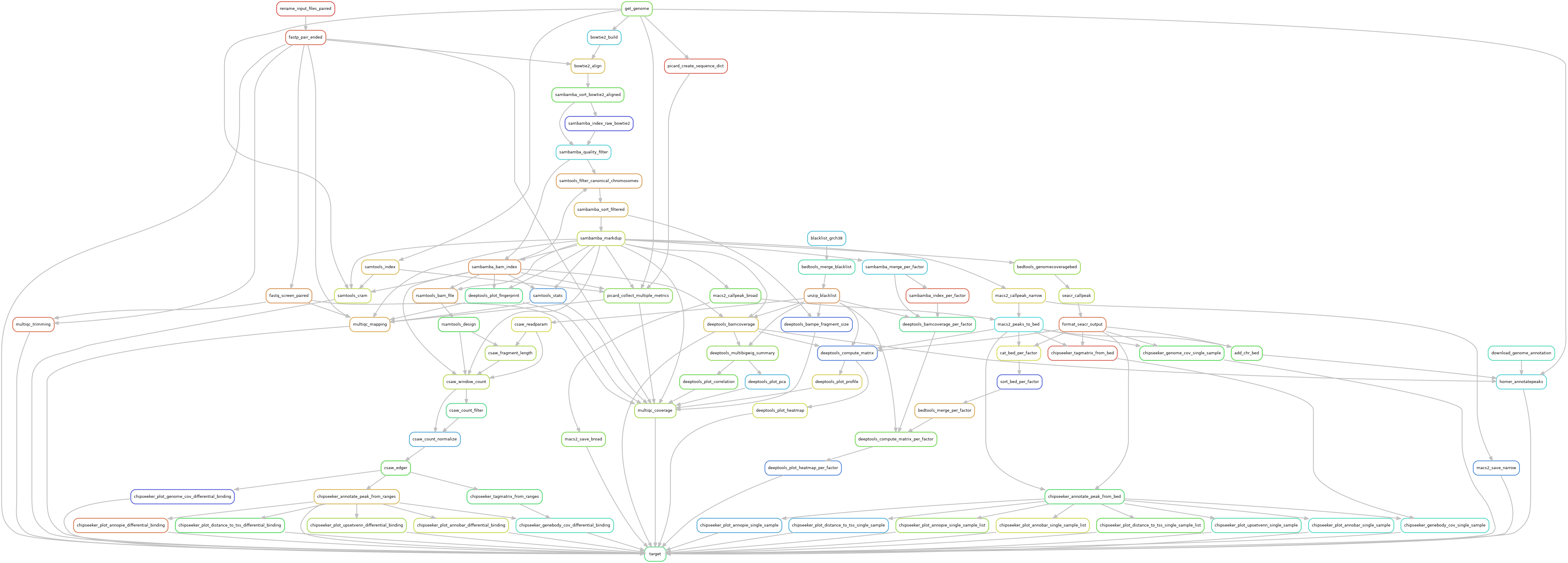
Reads are trimmed using
fastp
version
0.23.2
, using default parameters. Trimmed reads are mapped over the genome of interest defined in the configuration file, using
bowtie2
version
2.5.1
, using
--very-sensitive
parameter to increase mapping specificity.
Mapped reads with a quality lower than 30 are dropped out using
Sambamba
version
1.0
with parameter
--filter 'mapping_quality >= 30'
. In case of pair-ended library, orphan reads a dropped out using
--filter 'not (unmapped or mate_is_unmapped)'
in
Sambamba
version
1.0
. Remaining reads are filtered over canonical chromosomes, using
Samtools
version
1.17
. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome,
BedTools
version
2.31.0
with
-wa -sorted
as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were filtered out, using
Sambamba
version
1.0
using the optional parameter
--remove-duplicates --overflow-list-size 600000
.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0 , and Samtools version 1.17 .
Genome coverage was assessed with
DeepTools
version
3.5.2
. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. Gro-seq specificity was also taken into account using the following optional parameters
--normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY --Offset 12
. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by
Macs2
version
2.2.7.1
using both broad and narrow options. Pair-ended libraries recieved the
--format BAMPE
parameters, while single-ended libraries used estimated fragment size provided in input.
The peak annotation was done using ChIPSeeker version 1.34.0 , using Org.eg.db annotations version 3.16.0 .
De novo
motif discovery was made with
Homer
version
4.11
over called peaks, using the following optional parameters
-size 200 -homer2
over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed using
CSAW
version
1.32.0
. Input signal (if available) was used to find signal of interest over background noise. Or else, a large binning was defined over mappable genome regions to define a background noise track to use as an input signal. In any case, a log2 of 1.1 between the background and the sample of interest was used as a threshold value to identify regions of interest. Data were normalized over efficiency biases as described in
CSAW documentation
, since libraries of the same size can still have composition bias. Differential binded sites were called using EdgeR over the statistical formulas described in the configuration file. The
prior N
was defined over 20 degrees of freedom in the GLM test. FDR values from EdgeR were corrected using a region merge accross gene annotations to account for related binding sites. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3 . Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.0.0 .
Ribo-seq
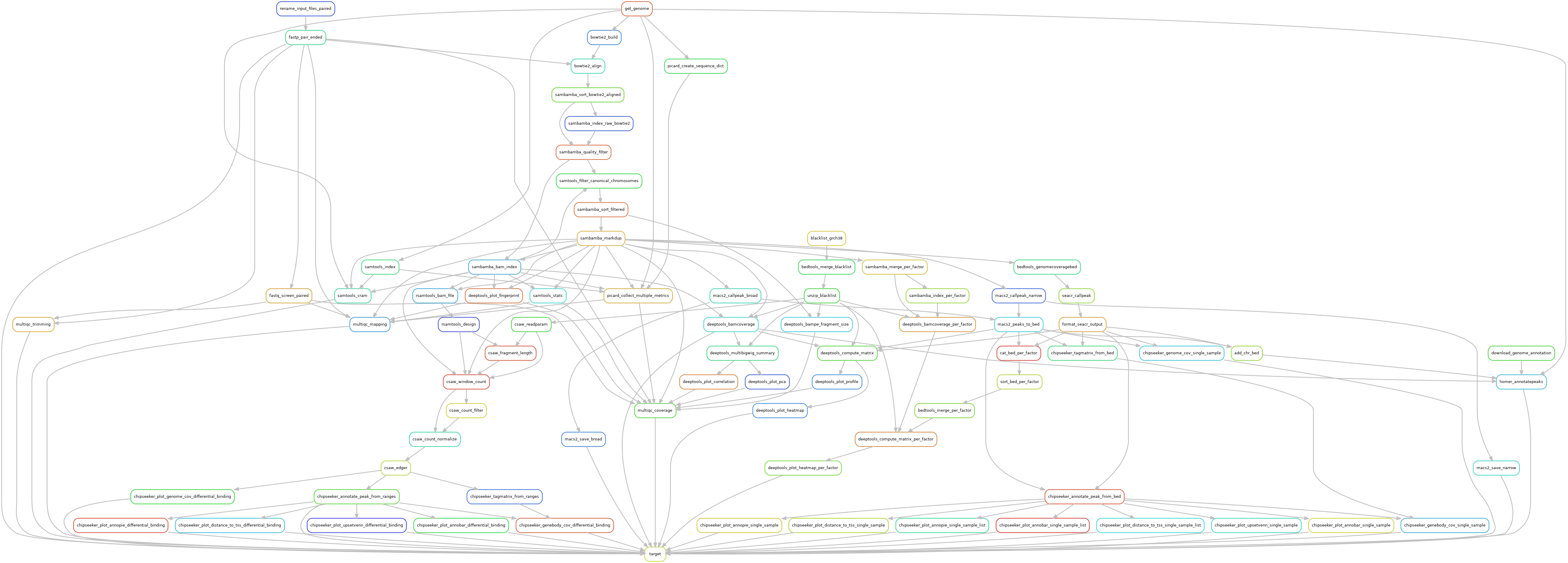
Reads are trimmed using
fastp
version
0.23.2
, using default parameters. Trimmed reads are mapped over the genome of interest defined in the configuration file, using
bowtie2
version
2.5.1
, using
--very-sensitive
parameter to increase mapping specificity.
Mapped reads with a quality lower than 30 are dropped out using
Sambamba
version
1.0
with parameter
--filter 'mapping_quality >= 30'
. In case of pair-ended library, orphan reads a dropped out using
--filter 'not (unmapped or mate_is_unmapped)'
in
Sambamba
version
1.0
. Remaining reads are filtered over canonical chromosomes, using
Samtools
version
1.17
. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome,
BedTools
version
2.31.0
with
-wa -sorted
as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were filtered out, using
Sambamba
version
1.0
using the optional parameter
--remove-duplicates --overflow-list-size 600000
.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0 , and Samtools version 1.17 .
Genome coverage was assessed with
DeepTools
version
3.5.2
. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. Ribo-seq specificity was also taken into account using the following optional parameters
--normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY --Offset 15
. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by
Macs2
version
2.2.7.1
using both broad and narrow options. Pair-ended libraries recieved the
--format BAMPE
parameters, while single-ended libraries used estimated fragment size provided in input.
The peak annotation was done using ChIPSeeker version 1.34.0 , using Org.eg.db annotations version 3.16.0 .
De novo
motif discovery was made with
Homer
version
4.11
over called peaks, using the following optional parameters
-size 200 -homer2
over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed using
CSAW
version
1.32.0
. Input signal (if available) was used to find signal of interest over background noise. Or else, a large binning was defined over mappable genome regions to define a background noise track to use as an input signal. In any case, a log2 of 1.1 between the background and the sample of interest was used as a threshold value to identify regions of interest. Data were normalized over efficiency biases as described in
CSAW documentation
, since libraries of the same size can still have composition bias. Differential binded sites were called using EdgeR over the statistical formulas described in the configuration file. The
prior N
was defined over 20 degrees of freedom in the GLM test. FDR values from EdgeR were corrected using a region merge accross gene annotations to account for related binding sites. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3 . Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.0.0 .
Atac-seq

Reads are trimmed using
fastp
version
0.23.2
, using default parameters. Trimmed reads are mapped over the genome of interest defined in the configuration file, using
bowtie2
version
2.5.1
, using
--very-sensitive
parameter to increase mapping specificity.
Mapped reads with a quality lower than 30 are dropped out using
Sambamba
version
1.0
with parameter
--filter 'mapping_quality >= 30'
. In case of pair-ended library, orphan reads a dropped out using
--filter 'not (unmapped or mate_is_unmapped)'
in
Sambamba
version
1.0
. Remaining reads are filtered over canonical chromosomes, using
Samtools
version
1.17
. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome,
BedTools
version
2.31.0
with
-wa -sorted
as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were filtered out, using
Sambamba
version
1.0
using the optional parameter
--remove-duplicates --overflow-list-size 600000
. Remaining reads were shifted as described in Buenrostro et al. 2013 : reads on the positive strand were shifted 4 bp to the right and reads on the negative were be shifted 5 bp to the left using
DeepTools
version
3.5.2
and the optional parameter
--ATACShift
.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0 , and Samtools version 1.17 .
Genome coverage was assessed with
DeepTools
version
3.5.2
. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. using the following optional parameters
--normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY
. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by
Macs2
version
2.2.7.1
using both broad and narrow options. Pair-ended libraries recieved the
--format BAMPE
parameters, while single-ended libraries used estimated fragment size provided in input.
The peak annotation was done using ChIPSeeker version 1.34.0 , using Org.eg.db annotations version 3.16.0 .
De novo
motif discovery was made with
Homer
version
4.11
over called peaks, using the following optional parameters
-size 200 -homer2
over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed using
CSAW
version
1.32.0
. A large binning was defined over mappable genome regions to define a background noise track to use as an input signal. In any case, a log2 of 1.1 between the background and the sample of interest was used as a threshold value to identify regions of interest. Data were normalized over efficiency biases as described in
CSAW documentation
, since libraries of the same size can still have composition bias. Differential binded sites were called using EdgeR over the statistical formulas described in the configuration file. The
prior N
was defined over 20 degrees of freedom in the GLM test. FDR values from EdgeR were corrected using a region merge accross gene annotations to account for related binding sites. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3 . Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.0.0 .
Cut&Tag

Reads are trimmed using
fastp
version
0.23.2
, using default parameters. Trimmed reads are mapped over the genome of interest defined in the configuration file, using
bowtie2
version
2.5.1
, using
--very-sensitive
parameter to increase mapping specificity.
Mapped reads with a quality lower than 30 are dropped out using
Sambamba
version
1.0
with parameter
--filter 'mapping_quality >= 30'
. In case of pair-ended library, orphan reads a dropped out using
--filter 'not (unmapped or mate_is_unmapped)'
in
Sambamba
version
1.0
. Remaining reads are filtered over canonical chromosomes, using
Samtools
version
1.17
. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome,
BedTools
version
2.31.0
with
-wa -sorted
as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were
not
filtered out, but marked as duplicates, using
Sambamba
version
1.0
using the optional parameter
--overflow-list-size 600000
.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0 , and Samtools version 1.17 .
Genome coverage was assessed with
DeepTools
version
3.5.2
. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. using the following optional parameters
--normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY --ignoreDuplicates
. Reads marked as duplicates were treated as normal reads. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by
Macs2
version
2.2.7.1
using both broad and narrow options. Pair-ended libraries recieved the
--format BAMPE
parameters, while single-ended libraries used estimated fragment size provided in input. Alongside with Macs2,
SEACR
version
1.3
was used to perform single-sample peak-calling using parameters hinted in the original publication of SEACR.
The peak annotation was done using ChIPSeeker version 1.34.0 , using Org.eg.db annotations version 3.16.0 .
De novo
motif discovery was made with
Homer
version
4.11
over called peaks, using the following optional parameters
-size 200 -homer2
over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed using
CSAW
version
1.32.0
. Input signal (if available) was used to find signal of interest over background noise. Or else, a large binning was defined over mappable genome regions to define a background noise track to use as an input signal. In any case, a log2 of 1.1 between the background and the sample of interest was used as a threshold value to identify regions of interest. Data were normalized over efficiency biases as described in
CSAW documentation
, since libraries of the same size can still have composition bias. Differential binded sites were called using EdgeR over the statistical formulas described in the configuration file. The
prior N
was defined over 20 degrees of freedom in the GLM test. FDR values from EdgeR were corrected using a region merge accross gene annotations to account for related binding sites. Through the whole process, reads flagged as duplicates are treated as any other read. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3 . Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.0.0 .
Cut&Run

Reads are trimmed using
fastp
version
0.23.2
, using default parameters. Trimmed reads are mapped over the genome of interest defined in the configuration file, using
bowtie2
version
2.5.1
, using
--very-sensitive
parameter to increase mapping specificity.
Mapped reads with a quality lower than 30 are dropped out using
Sambamba
version
1.0
with parameter
--filter 'mapping_quality >= 30'
. In case of pair-ended library, orphan reads a dropped out using
--filter 'not (unmapped or mate_is_unmapped)'
in
Sambamba
version
1.0
. Remaining reads are filtered over canonical chromosomes, using
Samtools
version
1.17
. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome,
BedTools
version
2.31.0
with
-wa -sorted
as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were
not
filtered out, but marked as duplicates, using
Sambamba
version
1.0
using the optional parameter
--overflow-list-size 600000
.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0 , and Samtools version 1.17 .
Genome coverage was assessed with
DeepTools
version
3.5.2
. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. using the following optional parameters
--normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY --ignoreDuplicates
. Reads marked as duplicates were treated as normal reads. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by
Macs2
version
2.2.7.1
using both broad and narrow options. Pair-ended libraries recieved the
--format BAMPE
parameters, while single-ended libraries used estimated fragment size provided in input.Alongside with Macs2,
SEACR
version
1.3
was used to perform single-sample peak-calling using parameters hinted in the original publication of SEACR.
The peak annotation was done using ChIPSeeker version 1.34.0 , using Org.eg.db annotations version 3.16.0 .
De novo
motif discovery was made with
Homer
version
4.11
over called peaks, using the following optional parameters
-size 200 -homer2
over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed using
CSAW
version
1.32.0
. Input signal (if available) was used to find signal of interest over background noise. Or else, a large binning was defined over mappable genome regions to define a background noise track to use as an input signal. In any case, a log2 of 1.1 between the background and the sample of interest was used as a threshold value to identify regions of interest. Data were normalized over efficiency biases as described in
CSAW documentation
, since libraries of the same size can still have composition bias. Differential binded sites were called using EdgeR over the statistical formulas described in the configuration file. The
prior N
was defined over 20 degrees of freedom in the GLM test. FDR values from EdgeR were corrected using a region merge accross gene annotations to account for related binding sites. Through the whole process, reads flagged as duplicates are treated as any other read. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3 . Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.0.0 .
MeDIP-Seq

Reads are trimmed using
fastp
version
0.23.2
, using default parameters. Trimmed reads are mapped over the genome of interest defined in the configuration file, using
bowtie2
version
2.5.1
, using
--very-sensitive
parameter to increase mapping specificity.
Mapped reads with a quality lower than 30 are dropped out using
Sambamba
version
1.0
with parameter
--filter 'mapping_quality >= 30'
. In case of pair-ended library, orphan reads a dropped out using
--filter 'not (unmapped or mate_is_unmapped)'
in
Sambamba
version
1.0
. Remaining reads are filtered over canonical chromosomes, using
Samtools
version
1.17
. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome,
BedTools
version
2.31.0
with
-wa -sorted
as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were filtered out, using
Sambamba
version
1.0
using the optional parameter
--remove-duplicates --overflow-list-size 600000
.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0 , and Samtools version 1.17 .
Genome coverage was assessed with
DeepTools
version
3.5.2
. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. using the following optional parameters
--normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY
. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by
Macs2
version
2.2.7.1
using both broad and narrow options. Pair-ended libraries recieved the
--format BAMPE
parameters, while single-ended libraries used estimated fragment size provided in input.
The peak annotation was done using ChIPSeeker version 1.34.0 , using Org.eg.db annotations version 3.16.0 .
De novo
motif discovery was made with
Homer
version
4.11
over called peaks, using the following optional parameters
-size 200 -homer2
over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed with MEDIPS version 1.50.0 . If available, input signals were used to search signal of interest over the background noise. An adjusted p-value threshold of 0.1 was chosen to find significant signal over noise. A distance of 1 base was used to merge neighboring significant windows. Differentially binded sites were called using EdgeR and FDR were corrected using MEDIPS internal methods as defined in the official documentation. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3 . Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.0.0 .
OxiDIP-Seq (OG-Seq)

Reads are trimmed using
fastp
version
0.23.2
, using the following non-default parameter:
--poly_g_min_len 25
since we expect a higher number of base modifications. Trimmed reads are mapped over the genome of interest defined in the configuration file, using
bowtie2
version
2.5.1
, using default parameters.
Mapped reads with a quality lower than 30 are dropped out using
Sambamba
version
1.0
with parameter
--filter 'mapping_quality >= 30'
. In case of pair-ended library, orphan reads a dropped out using
--filter 'not (unmapped or mate_is_unmapped)'
in
Sambamba
version
1.0
. Remaining reads are filtered over canonical chromosomes, using
Samtools
version
1.17
. Mitochondrial chromosome is not considered as a canonical chromosome, that being so, mitochondrial reads are dropped out. In case regions of interest are defined over the genome,
BedTools
version
2.31.0
with
-wa -sorted
as optional parameter to keep mapped reads that overlap the regions of interest by, at least, one base. Duplicated reads were filtered out, using
Sambamba
version
1.0
using the optional parameter
--remove-duplicates --overflow-list-size 600000
. GC bias was estimated and corrected using
DeepTools
version
3.5.2
using default parameters.
A quality control is made both before and after these filters to ensure no valuable data is lost. These quality controls are made using Picard version 3.0.0 , and Samtools version 1.17 .
Genome coverage was assessed with
DeepTools
version
3.5.2
. Chromosome X and Y were ignored in the RPKM normalization in order to reduce noise the final result. using the following optional parameters
--normalizeUsing RPKM --binSize 5 --skipNonCoveredRegions --ignoreForNormalization chrX chrM chrY
. The same tool was used to plot both quality controls and peak coverage heatmaps.
Single-sample peak calling was performed by
Macs2
version
2.2.7.1
using both broad and narrow options. Pair-ended libraries recieved the
--format BAMPE
parameters, while single-ended libraries used estimated fragment size provided in input.
The peak annotation was done using ChIPSeeker version 1.34.0 , using Org.eg.db annotations version 3.16.0 .
De novo
motif discovery was made with
Homer
version
4.11
over called peaks, using the following optional parameters
-size 200 -homer2
over the genome identified in the configuration file (GRCh38 or mm10). The motifs were used for further annotation of peaks with Homer.
Differential binding analysis was performed using
CSAW
version
1.32.0
. Input signal (if available) was used to find signal of interest over background noise. Or else, a large binning was defined over mappable genome regions to define a background noise track to use as an input signal. In any case, a log2 of 1.1 between the background and the sample of interest was used as a threshold value to identify regions of interest. Data were normalized over efficiency biases as described in
CSAW documentation
, since libraries of the same size can still have composition bias, and 8oxoG changes the base composition. Differential binded sites were called using EdgeR over the statistical formulas described in the configuration file. The
prior N
was defined over 20 degrees of freedom in the GLM test. FDR values from EdgeR were corrected using a region merge accross gene annotations to account for related binding sites. Through the whole process, reads flagged as duplicates are treated as any other read. Differentially bounded regions were annotated as if they were peaks, using the same methods, and the same parameters.
Additional quality controls were made using FastqScreen version 0.15.3 . Complete quality reports were built using MultiQC aggregation tool and in-house scripts.
The whole pipeline was powered by Snakemake version 7.26.0 and the Snakemake-Wrappers version v2.0.0 .
Citations
-
Fastp:
Chen, Shifu, et al. "fastp: an ultra-fast all-in-one FASTQ preprocessor." Bioinformatics 34.17 (2018): i884-i890. -
Bowtie2:
Langmead, Ben, and Steven L. Salzberg. "Fast gapped-read alignment with Bowtie 2." Nature methods 9.4 (2012): 357-359. -
Sambamba:
Tarasov, Artem, et al. "Sambamba: fast processing of NGS alignment formats." Bioinformatics 31.12 (2015): 2032-2034. -
Ensembl:
Martin, Fergal J., et al. "Ensembl 2023." Nucleic Acids Research 51.D1 (2023): D933-D941. -
Snakemake:
Köster, Johannes, and Sven Rahmann. "Snakemake—a scalable bioinformatics workflow engine." Bioinformatics 28.19 (2012): 2520-2522. -
Atac-Seq:
Buenrostro, Jason D., et al. "Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position." Nature methods 10.12 (2013): 1213-1218. -
MeDIPS:
Lienhard, Matthias, et al. "MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments." Bioinformatics 30.2 (2014): 284-286. -
DeepTools:
Ramírez, Fidel, et al. "deepTools: a flexible platform for exploring deep-sequencing data." Nucleic acids research 42.W1 (2014): W187-W191. -
UCSC:
Nassar LR, Barber GP, Benet-Pagès A, Casper J, Clawson H, Diekhans M, Fischer C, Gonzalez JN, Hinrichs AS, Lee BT, Lee CM, Muthuraman P, Nguy B, Pereira T, Nejad P, Perez G, Raney BJ, Schmelter D, Speir ML, Wick BD, Zweig AS, Haussler D, Kuhn RM, Haeussler M, Kent WJ. The UCSC Genome Browser database: 2023 update. Nucleic Acids Res. 2022 Nov 24;. PMID: 36420891 -
ChIPSeeker:
Yu, Guangchuang, Li-Gen Wang, and Qing-Yu He. "ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization." Bioinformatics 31.14 (2015): 2382-2383. -
CSAW:
Lun, Aaron TL, and Gordon K. Smyth. "csaw: a Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows." Nucleic acids research 44.5 (2016): e45-e45. -
Macs2:
Gaspar, John M. "Improved peak-calling with MACS2." BioRxiv (2018): 496521. -
Homer:
Heinz S, Benner C, Spann N, Bertolino E et al. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol Cell 2010 May 28;38(4):576-589. PMID: 20513432 -
Seacr:
Meers, Michael P., Dan Tenenbaum, and Steven Henikoff. "Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling."Epigenetics & chromatin 12 (2019): 1-11. -
BedTools:
Quinlan, Aaron R., and Ira M. Hall. "BEDTools: a flexible suite of utilities for comparing genomic features." Bioinformatics 26.6 (2010): 841-842. -
Snakemake-Wrappers version v2.0.0
Roadmap
-
Coverage: PBC
-
Peak-annotation: CentriMo
-
Differential Peak Calling: DiffBind
-
IGV: screen-shot, igv-reports
Based on Snakemake-Wrappers version v2.0.0
Code Snippets
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 | __author__ = "Thibault Dayris" __copyright__ = "Copyright 2023, Thibault Dayris" __email__ = "[email protected]" __license__ = "MIT" from snakemake.shell import shell log = snakemake.log_fmt_shell(stdout=True, stderr=True) extra = snakemake.params.get("extra", "") blacklist = snakemake.input.get("blacklist", "") if blacklist: extra += f" --blackListFileName {blacklist} " out_file = snakemake.output[0] if out_file.endswith(".bed"): extra += " --BED " shell( "alignmentSieve " "{extra} " "--numberOfProcessors {snakemake.threads} " "--bam {snakemake.input.aln} " "--outFile {out_file} " "{log} " ) |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 | __author__ = "Jan Forster, Felix Mölder" __copyright__ = "Copyright 2019, Jan Forster" __email__ = "[email protected], [email protected]" __license__ = "MIT" from snakemake.shell import shell ## Extract arguments extra = snakemake.params.get("extra", "") log = snakemake.log_fmt_shell(stdout=True, stderr=True) if len(snakemake.input) > 1: if all(f.endswith(".gz") for f in snakemake.input): cat = "zcat" elif all(not f.endswith(".gz") for f in snakemake.input): cat = "cat" else: raise ValueError("Input files must be all compressed or uncompressed.") shell( "({cat} {snakemake.input} | " "sort -k1,1 -k2,2n | " "bedtools merge {extra} " "-i stdin > {snakemake.output}) " " {log}" ) else: shell( "( bedtools merge" " {extra}" " -i {snakemake.input}" " > {snakemake.output})" " {log}" ) |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 | __author__ = "Jan Forster" __copyright__ = "Copyright 2021, Jan Forster" __email__ = "[email protected]" __license__ = "MIT" import os from snakemake.shell import shell log = snakemake.log_fmt_shell(stdout=True, stderr=True) shell( "sambamba index {snakemake.params.extra} -t {snakemake.threads} " "{snakemake.input[0]} {snakemake.output[0]} " "{log}" ) |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 | __author__ = "Jan Forster" __copyright__ = "Copyright 2021, Jan Forster" __email__ = "[email protected]" __license__ = "MIT" import os from snakemake.shell import shell log = snakemake.log_fmt_shell(stdout=True, stderr=True) shell( "sambamba merge {snakemake.params.extra} -t {snakemake.threads} " "{snakemake.output[0]} {snakemake.input} " "{log}" ) |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 | __author__ = "Jan Forster" __copyright__ = "Copyright 2021, Jan Forster" __email__ = "[email protected]" __license__ = "MIT" import os from snakemake.shell import shell in_file = snakemake.input[0] extra = snakemake.params.get("extra", "") log = snakemake.log_fmt_shell(stdout=False, stderr=True) if in_file.endswith(".sam") and ("-S" not in extra or "--sam-input" not in extra): extra += " --sam-input" shell( "sambamba view {extra} -t {snakemake.threads} " "{snakemake.input[0]} > {snakemake.output[0]} " "{log}" ) |
18 19 | script: "../../scripts/chipseeker/chipseeker_plot_gene_body.R" |
39 40 | script: "../../scripts/chipseeker/chipseeker_plot_gene_body.R" |
17 18 | script: "../../scripts/chipseeker/chipseeker_covplot.R" |
37 38 | script: "../../scripts/chipseeker/chipseeker_covplot.R" |
18 19 | script: "../../scripts/chipseeker/chipseeker_annotate.R" |
44 45 | script: "../../scripts/chipseeker/chipseeker_annotate.R" |
22 23 | script: "../../scripts/chipseeker/chipseeker_plot_annobar.R" |
42 43 | script: "../../scripts/chipseeker/chipseeker_plot_annobar.R" |
66 67 | script: "../../scripts/chipseeker/chipseeker_plot_annobar_lsit.R" |
22 23 | script: "../../scripts/chipseeker/chipseeker_plot_distance_tss.R" |
42 43 | script: "../../scripts/chipseeker/chipseeker_plot_distance_tss.R" |
66 67 | script: "../../scripts/chipseeker/chipseeker_plot_distance_tss_list.R" |
17 18 | script: "../../scripts/chipseeker/chipseeker_plot_upsetvenn.R" |
37 38 | script: "../../scripts/chipseeker/chipseeker_plot_upsetvenn.R" |
17 18 | script: "../../scripts/chipseeker/chipseeker_make_tagmatrix.R" |
37 38 | script: "../../scripts/chipseeker/chipseeker_make_tagmatrix.R" |
19 20 21 22 23 24 25 26 27 | shell: "annotatePeaks.pl " "{input.peak} " "{params.genome} " "-cpu {threads} " "-wig {input.wig} " "{params.extra} " "> {output.annotations} " "2> {log} " |
17 18 | script: "../../scripts/csaw/csaw_fregment_length.R" |
37 38 | script: "../../scripts/csaw/csaw_count.R" |
63 64 | script: "../../scripts/csaw/csaw_filter.R" |
41 42 43 44 45 46 47 | shell: "multiBigwigSummary bins {params.extra} " "--bwfiles {input.bw} " "--outFileName {output.bw} " "--blackListFileName {input.blacklist} " "--numberOfProcessors {threads} " "> {log} 2>&1 " |
17 18 | script: "../../scripts/medip-seq/medips_meth.R" |
18 19 20 21 22 | shell: "plotCorrelation --corData {input.bw} " "--plotFile {output.png} " "--outFileCorMatrix {output.stats} " "{params.extra} > {log} 2>&1 " |
18 19 20 21 22 | shell: "plotPCA {params.extra} --corData {input.bw} " "--plotFile {output.png} " "--outFileNameData {output.stats} " "> {log} 2>&1 " |
20 21 | script: "../../scripts/csaw/csaw_normalize.R" |
46 47 | script: "../../scripts/csaw/csaw_edger.R" |
17 18 | script: "../../scripts/medip-seq/medips_edger.R" |
15 16 | wrapper: "v2.0.0/bio/sambamba/merge" |
19 20 | shell: "cat {params} {input} > {output} 2> {log}" |
41 42 | shell: "sort {params} {input} > {output} 2> {log}" |
59 60 | wrapper: "v2.0.0/bio/bedtools/merge" |
22 23 | script: "../../scripts/factorfootprints/factor_footprints.R" |
47 48 | script: "../../scripts/factorfootprints/factor_footprints.R" |
18 19 | script: "../../scripts/csaw/csaw_readparam.R" |
15 16 | script: "../../scripts/misc/rename.py" |
33 34 | script: "../../scripts/misc/rename.py" |
31 32 33 34 35 36 37 38 39 40 | shell: "xenome index {params.extra} " "--num-threads {threads} " "--max-memory {resources.mem_mb} " "--prefix {params.prefix} " "--graft {input.human} " "--host {input.mouse} " "--log-file {log.tool} " "--tmp-dir {resources.tmpdir} " "> {log.general} 2>&1 " |
19 20 | shell: "cut {params.cut} {input} | sort {params.sort} > {output} 2> {log}" |
39 40 41 42 43 44 45 | shell: "bedtools intersect " "{params.extra} " "-g {input.genome} " "-abam {input.left} " "-b {input.right} " "> {output} 2> {log}" |
22 23 24 25 26 27 28 29 30 31 32 | shell: "computeGCBias " "--bamfile {input.bam} " "--genome {input.genome} " "--GCbiasFrequenciesFile {output.freq} " "--effectiveGenomeSize {params.genome_size} " "--blackListFileName {input.blacklist} " "--numberOfProcessors {threads} " "--biasPlot {output.plot} " "{params.extra} " "> {log} 2>&1 " |
55 56 57 58 59 60 61 62 63 64 | shell: "correctGCBias " "--bamfile {input.bam} " "--genome {input.genome} " "--GCbiasFrequenciesFile {input.freq} " "--correctedFile {output} " "--effectiveGenomeSize {params.genome_size} " "--numberOfProcessors {threads} " "{params.extra} " "> {log} 2>&1 " |
17 18 | wrapper: "master/bio/deeptools/alignmentsieve" |
73 74 | wrapper: "master/bio/deeptools/alignmentsieve" |
17 18 | wrapper: "v2.0.0/bio/sambamba/view" |
39 40 | shell: "sed {params} {input} > {output} 2> {log}" |
59 60 | wrapper: "v2.0.0/bio/sambamba/view" |
81 82 | shell: "awk --file {params.awk} {input} > {output} 2> {log}" |
103 104 | shell: "sed {params} {input} > {output} 2> {log}" |
126 127 | shell: "cat {params.extra} {input.header} {input.reads} > {output} 2> {log}" |
146 147 | wrapper: "v2.0.0/bio/sambamba/view" |
182 183 | wrapper: "v2.0.0/bio/sambamba/index" |
202 203 | wrapper: "v2.0.0/bio/sambamba/view" |
224 225 | shell: "sed {params} {input} > {output} 2> {log}" |
244 245 | wrapper: "v2.0.0/bio/sambamba/view" |
266 267 | shell: "sed {params} {input} > {output} 2> {log}" |
288 289 | shell: "awk --file {params.awk} {input} > {output} 2> {log}" |
311 312 | shell: "cat {params.extra} {input.header} {input.reads} > {output} 2> {log}" |
331 332 | wrapper: "v2.0.0/bio/sambamba/view" |
367 368 | wrapper: "v2.0.0/bio/sambamba/index" |
15 16 | wrapper: "v2.0.0/bio/sambamba/merge" |
33 34 | wrapper: "v2.0.0/bio/sambamba/index" |
22 23 24 25 26 27 28 29 | shell: "bamPEFragmentSize " "{params.extra} " "--bamfiles {input.bam} " "--histogram {output.hist} " "--numberOfProcessors {threads} " "--blackListFileName {input.blacklist} " "> {output.metrics} 2> {log} " |
15 16 | script: "../../scripts/rsamtools/create_bam_file.R" |
35 36 | script: "../../scripts/rsamtools/create_design.R" |
27 28 29 30 31 32 33 34 35 36 | shell: "xenome classify {params.extra} " "--log-file {log.tool} " "--num-threads {threads} " "--tmp-dir {resources.tmpdir} " "--max-memory {resources.mem_mb} " "--output-filename-prefix {params.out_prefix} " "--prefix {params.idx_prefix} " "--fastq-in {input.r1} --fastq-in {input.r2} " "> {log.general} 2>&1 " |
64 65 66 67 68 69 70 71 72 73 | shell: "xenome classify {params.extra} " "--log-file {log.tool} " "--num-threads {threads} " "--tmp-dir {resources.tmpdir} " "--max-memory {resources.mem_mb} " "--output-filename-prefix {params.out_prefix} " "--prefix {params.idx_prefix} " "--fastq-in {input.reads} " "> {log.general} 2>&1 " |
17 18 | script: "../../scripts/medips_load.R" |
37 38 | script: "../../scripts/medips_coupling.R" |
17 18 | shell: "awk --file {params.script} {input} > {output} 2> {log}" |
40 41 42 43 44 45 46 47 48 | shell: "findMotifsGenome.pl " "{input.peak} " "{params.genome} " "{params.output_directory} " "-size {params.size} " "{params.extra} " "-p {threads} " "> {log} 2>&1 " |
37 38 | shell: "rsync {params} {input} {output} > {log} 2>&1" |
77 78 | shell: "rsync {params} {input} {output} > {log} 2>&1" |
98 99 | shell: "awk --file {params.script} {input} > {output} 2> {log}" |
17 18 19 | shell: "NB=$(sambamba view {params} {input}) && " 'echo -e "{wildcards.sample}\t${{NB}}" > {output} 2> {log}' |
38 39 40 | shell: "NB=$(sambamba view {params} {input}) && " 'echo -e "{wildcards.sample}\t${{NB}}" > {output} 2> {log}' |
64 65 | shell: "paste - - <(cat {input.called}) <(cat {input.total}) > {output} 2> {log}" |
84 85 | shell: "awk --file {params.script} {input} > {output} 2> {log}" |
35 36 37 38 39 | shell: "SEACR_1.3.sh " "{input.exp_bg} " "{params.extra} " "> {log} 2>&1" |
58 59 | shell: "awk --file {params.script} {input} > {output} 2> {log}" |
18 19 | shell: "wget {params.extra} {params.address} -O {output} > {log} 2>&1" |
39 40 | shell: "wget {params.extra} {params.address} -O {output} > {log} 2>&1" |
60 61 | shell: "wget {params.extra} {params.address} -O {output} > {log} 2>&1" |
81 82 | shell: "wget {params.extra} {params.address} -O {output} > {log} 2>&1" |
21 22 23 | shell: "wget {params} --directory-prefix {output} " "{params.url} > {log} 2>&1" |
46 47 48 | shell: "wget {params} --directory-prefix {output} " "{params.url} > {log} 2>&1" |
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 | log_file <- file(snakemake@log[[1]], open = "wt") sink(log_file) sink(log_file, type = "message") # Load libraries base::library(package = "ChIPseeker", character.only = TRUE) base::library(package = "org.Hs.eg.db", character.only = TRUE) base::library(package = "org.Mm.eg.db", character.only = TRUE) base::library(package = "EnsDb.Hsapiens.v86", character.only = TRUE) base::library(package = "EnsDb.Mmusculus.v79", character.only = TRUE) base::message("Libraries loaded") organism <- "hg38" if ("organism" %in% base::names(x = snakemake@params)) { organism <- base::as.character(x = snakemake@params[["organism"]]) } organism <- base::tolower(x = organism) if (organism == "hg38") { anno_db <- "org.Hs.eg.db" edb <- EnsDb.Hsapiens.v86 } else if (organism == "mm10") { anno_db <- "org.Mm.eg.db" edb <- EnsDb.Mmusculus.v79 } else { base::stop("Unknown organism annotation") } seqlevelsStyle(edb) <- "Ensembl" ranges <- NULL if ("ranges" %in% base::names(x = snakemake@input)) { ranges <- base::readRDS(file = base::as.character(x = snakemake@input[["ranges"]])) } else { ranges <- ChIPseeker::readPeakFile( peakfile = base::as.character(x = snakemake@input[["bed"]]), as = "GRanges" ) } base::message("Parameters and data loaded") # Building command line extra <- "peak = ranges, annoDb = anno_db, TxDb = edb" if ("extra" %in% base::names(x = snakemake@params)) { extra <- base::paste( extra, base::as.character(x = snakemake@params[["extra"]]), sep = ", " ) } command <- base::paste0( "ChIPseeker::annotatePeak(", extra, ")" ) base::message("Command line used:") base::message(command) # Annotating annotation <- base::eval(base::parse(text = command)) base::message("Data annotated") # Saving results base::saveRDS( object = annotation, file = base::as.character(x = snakemake@output[["rds"]]) ) base::message("RDS saved") utils::write.table( base::as.data.frame(annotation), sep = "\t", col.names = TRUE, row.names = TRUE, file = base::as.character(x = snakemake@output["tsv"]) ) base::message("Provess over") # Proper syntax to close the connection for the log file # but could be optional for Snakemake wrapper base::sink(type = "message") base::sink() |
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 | log_file <- file(snakemake@log[[1]], open = "wt") sink(log_file) sink(log_file, type = "message") base::message("Logging defined") # Load libraries base::library(package = "ChIPseeker", character.only = TRUE) base::message("Libraries loaded") peak <- NULL if ("bed" %in% base::names(x = snakemake@input)) { peak <- ChIPseeker::readPeakFile( peakfile = base::as.character(x = snakemake@input[["bed"]]), as = "GRanges", header = FALSE ) } else { peak <- base::readRDS( file = base::as.character(x = snakemake@input[["ranges"]]) ) } base::message("Ranges loaded") # Build plot grDevices::png( filename = snakemake@output[["png"]], width = 1024, height = 768, units = "px", type = "cairo" ) ChIPseeker::covplot(peak, weightCol = "V5") dev.off() base::message("Plot saved") # Proper syntax to close the connection for the log file # but could be optional for Snakemake wrapper base::sink(type = "message") base::sink() |
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 | log_file <- file(snakemake@log[[1]], open = "wt") sink(log_file) sink(log_file, type = "message") base::message("Logging defined") # Load libraries base::library(package = "ChIPseeker", character.only = TRUE) base::library( package = "TxDb.Hsapiens.UCSC.hg38.knownGene", character.only = TRUE ) base::library( package = "TxDb.Mmusculus.UCSC.mm10.knownGene", character.only = TRUE ) base::message("Libraries loaded") # Load peaks if ("bed" %in% base::names(x = snakemake@input)) { peaks <- base::as.character(x = snakemake@input[["bed"]]) } else if ("ranges" %in% base::names(x = snakemake@input)) { peaks <- base::readRDS( file = base::as.character(x = snakemake@input[["ranges"]]) ) } base::message("Peaks loaded") # Select transcript database txdb <- TxDb.Hsapiens.UCSC.hg38.knownGene if ("organism" %in% base::names(x = snakemake@params)) { if (snakemake@params[["organism"]] == "mm10") { organism <- TxDb.Mmusculus.UCSC.mm10.knownGene } } seqlevelsStyle(txdb) <- "Ensembl" # Acquire promoter position promoter <- ChIPseeker::getPromoters( TxDb = txdb, upstream = 3000, downstream = 3000, by = 'gene' ) # Build tagmatrix tagMatrix <- ChIPseeker::getTagMatrix( peak = peaks, windows = promoter ) base::message("TagMatrix built") # Save object base::saveRDS( file = base::as.character(x = snakemake@output[["rds"]]), object = tagMatrix ) # Proper syntax to close the connection for the log file # but could be optional for Snakemake wrapper base::sink(type = "message") base::sink() base::message("Process over") |
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 | log_file <- file(snakemake@log[[1]], open = "wt") sink(log_file) sink(log_file, type = "message") base::message("Logging defined") # Load libraries base::library(package = "ChIPseeker", character.only = TRUE) base::message("Libraries loaded") ranges <- base::readRDS( file = base::as.character(x = snakemake@input[["ranges"]]) ) base::message("Ranges loaded") # Build plot grDevices::png( filename = snakemake@output[["png"]], width = 1024, height = 384, units = "px", type = "cairo" ) ChIPseeker::plotAnnoBar(ranges) dev.off() base::message("Plot saved") # Proper syntax to close the connection for the log file # but could be optional for Snakemake wrapper base::sink(type = "message") base::sink() |
11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 | log_file <- file(snakemake@log[[1]], open = "wt") sink(log_file) sink(log_file, type = "message") base::message("Logging defined") # Load libraries base::library(package = "ChIPseeker", character.only = TRUE) base::message("Libraries loaded") ranges_list <- base::lapply( snakemake@input[["ranges"]], function(range_path) { base::readRDS( file = base::as.character(x = range_path) ) } ) base::message("Ranges loaded") # Build plot grDevices::png( filename = snakemake@output[["png"]], width = 1024, height = 384, units = "px", type = "cairo" ) ChIPseeker::plotDistToTSS(ranges_list) dev.off() base::message("Plot saved") # Proper syntax to close the connection for the log file # but could be optional for Snakemake wrapper base::sink(type = "message") base::sink() |
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 | log_file <- file(snakemake@log[[1]], open = "wt") sink(log_file) sink(log_file, type = "message") base::message("Logging defined") # Load libraries base::library(package = "ChIPseeker", character.only = TRUE) base::message("Libraries loaded") ranges <- base::readRDS( file = base::as.character(x = snakemake@input[["ranges"]]) ) base::message("Ranges loaded") # Build plot grDevices::png( filename = snakemake@output[["png"]], width = 1024, height = 384, units = "px", type = "cairo" ) ChIPseeker::plotDistToTSS(ranges) dev.off() base::message("Plot saved") # Proper syntax to close the connection for the log file # but could be optional for Snakemake wrapper base::sink(type = "message") base::sink() |
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 | log_file <- file(snakemake@log[[1]], open = "wt") sink(log_file) sink(log_file, type = "message") base::message("Logging defined") # Load libraries base::library(package = "ChIPseeker", character.only = TRUE) base::library( package = "TxDb.Hsapiens.UCSC.hg38.knownGene", character.only = TRUE ) base::library( package = "TxDb.Mmusculus.UCSC.mm10.knownGene", character.only = TRUE ) ncpus <- base::as.numeric(snakemake@threads[[1]]) mc.cores <- ncpus base::message("Libraries loaded") ranges <- NULL if ("bed" %in% base::names(x = snakemake@input)) { bed_path <- base::as.character(x = snakemake@input[["bed"]]) ranges <- ChIPseeker::readPeakFile( peakfile = bed_path, as = "GRanges", header = FALSE ) } else if ("ranges" %in% base::names(x = snakemake@input)) { ranges <- base::readRDS( file = base::as.character(x = snakemake@input[["ranges"]]) ) } base::message("Peaks/Ranges loaded") tag_matrix <- base::readRDS( file = base::as.character(x = snakemake@input[["tagmatrix"]]) ) upstream <- base::attr(x = tag_matrix, which = "upsteam") downstream <- base::attr(x = tag_matrix, which = "downstream") base::message("Tagmatrix loaded") # The default resample value resample <- 500 window_number <- base::length(tag_matrix) # Search all prime factors for the given number of windows prime_factors <- function(x) { factors <- c() last_prime <- 2 while(x >= last_prime){ if (! x %% last_prime) { factors <- c(factors, last_prime) x <- x / last_prime last_prime <- last_prime - 1 } last_prime <- last_prime + 1 } base::return(factors) } primes <- base::as.data.frame(x = base::table(prime_factors(x = window_number))) primes$Var1 <- base::as.integer(base::as.character(primes$Var1)) primes <- primes$Var1 ^ primes$Freq # Closest prime to default resample value primes_min <- base::abs(primes - resample) resample <- primes[base::which(primes_min == base::min(primes_min))] base::message("Resampling value: ", resample) txdb <- TxDb.Hsapiens.UCSC.hg38.knownGene if ("organism" %in% base::names(x = snakemake@params)) { if (snakemake@params[["organism"]] == "mm10") { organism <- TxDb.Mmusculus.UCSC.mm10.knownGene } } base::message("Organism library available") # Build plot grDevices::png( filename = snakemake@output[["png"]], width = 1024, height = 768, units = "px", type = "cairo" ) ChIPseeker::plotPeakProf( peak = ranges, tagMatrix = tag_matrix, upstream = upstream, # rel(0.2), downstream = downstream, # rel(0.2), weightCol = "V5", ignore_strand = TRUE, conf = 0.95, by = "gene", type = "body", TxDb = txdb, facet = "row", nbin = 800, verbose = TRUE, resample = resample ) dev.off() base::message("Plot saved") # Proper syntax to close the connection for the log file # but could be optional for Snakemake wrapper base::sink(type = "message") base::sink() |
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 | log_file <- file(snakemake@log[[1]], open = "wt") sink(log_file) sink(log_file, type = "message") base::message("Logging defined") # Load libraries base::library(package = "ChIPseeker", character.only = TRUE) base::message("Libraries loaded") ranges <- base::readRDS( file = base::as.character(x = snakemake@input[["ranges"]]) ) base::message("Ranges loaded") # Build plot grDevices::png( filename = snakemake@output[["png"]], width = 1024, height = 768, units = "px", type = "cairo" ) ChIPseeker::upsetplot(ranges, vennpie = TRUE) dev.off() base::message("Plot saved") # Proper syntax to close the connection for the log file # but could be optional for Snakemake wrapper base::sink(type = "message") base::sink() |
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 | log_file <- file(snakemake@log[[1]], open = "wt") sink(log_file) sink(log_file, type = "message") # Load libraries base::library(package = "BiocParallel", character.only = TRUE) base::library(package = "csaw", character.only = TRUE) base::message("Libraries loaded") if (snakemake@threads > 1) { BiocParallel::register( BiocParallel::MulticoreParam(snakemake@threads) ) options("mc.cores" = snakemake@threads) base::message("Process multi-threaded") } # Loading list of input files design <- readRDS( file = base::as.character(x = snakemake@input[["design"]]) ) frag_length <- readRDS( file = base::as.character(x = snakemake@input[["fragment_length"]]) ) # Loading filter parameters read_params <- base::readRDS( file = base::as.character(x = snakemake@input[["read_params"]]) ) base::message("Input data loaded") extra <- "bam.files = design$BamPath, param = read_params, ext=frag_length" if ("extra" %in% base::names(x = snakemake@params)) { extra <- base::paste( extra, base::as.character(x = snakemake@params[["extra"]]), sep = ", " ) } command <- base::paste( "csaw::windowCounts(", extra, ")" ) base::message("Count command line:") base::print(command) # Counting reads over sliding window counts <- base::eval(base::parse(text = command)) base::message( "Number of extended reads ", "overlapping a sliding window ", "at spaced positions across the ", "genome, acquired." ) # Saving results base::saveRDS( object = counts, file = snakemake@output[["rds"]] ) base::print(counts$totals) base::message("RDS saved") if ("ranges" %in% base::names(snakemake@output)) { if (base::endsWith(x = snakemake@output[["ranges"]], suffix = ".RDS")) { base::saveRDS( object = rowRanges(x = counts), file = snakemake@output[["ranges"]] ) } else { utils::write.table( x = base::as.data.frame(rowRanges(x = counts)), file = snakemake@output[["ranges"]], sep = "\t", col.names = TRUE, row.names = TRUE ) } } # base::message("Total number of ranges counted: ") # base::print(counts$totals) base::message("Process over.") # Proper syntax to close the connection for the log file # but could be optional for Snakemake wrapper base::sink(type = "message") base::sink() |
11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 | log_file <- file(snakemake@log[[1]], open = "wt") sink(log_file) sink(log_file, type = "message") # Load libraries base::library(package = "edgeR", character.only = TRUE) base::library(package = "csaw", character.only = TRUE) base::message("Libraries loaded") counts <- base::readRDS( file = base::as.character(x = snakemake@input[["counts"]]) ) y <- csaw::asDGEList(object = counts) base::message("Normalized-filtered counts loaded") exp_design <- utils::read.table( file = base::as.character(x = snakemake@input[["design"]]), sep = "\t", header = TRUE, stringsAsFactors = FALSE ) design <- stats::model.matrix( object = stats::as.formula(snakemake@params[["formula"]]), data = exp_design ) base::message("Model matrix built") prior.n <- edgeR::getPriorN( y = y, design = design, prior.df = 20 ) base::message("Prior N estimated with 20 dof") y <- edgeR::estimateDisp( y = y, design = design, prior.n = prior.n, ) base::message("Dispersion estimated") grDevices::png( filename = base::as.character(x = snakemake@output[["disp_png"]]), width = 1024, height = 768 ) edgeR::plotBCV(y = y) grDevices::dev.off() base::message("Dispersion plot saved") fit <- edgeR::glmQLFit( y = y, design = design, robust = TRUE ) base::message("GLM model fitted") results <- edgeR::glmQLFTest( glmfit = fit, coef = utils::tail(x = base::colnames(x = design), 1) ) base::message("Window counts tested") grDevices::png( filename = base::as.character(x = snakemake@output[["ql_png"]]), width = 1024, height = 768 ) edgeR::plotQLDisp(glmfit = results) grDevices::dev.off() base::message("Quasi-likelihood dispersion plot saved") # rowData(counts) <- base::cbind(rowData(counts), results$table) merged <- csaw::mergeResults( counts, results$table, tol = 100, merge.args = list(max.width = 5000) ) base::message("Results stored in RangedSummarizedExperiment object") # Save results if ("qc" %in% base::names(snakemake@output)) { is_significative <- summary(merged$combined$FDR <= 0.05) direction <- summary(table(merged$combined$direction[is_significative])) sig <- base::data.frame( Differentially_Expressed = is_significative[["TRUE"]], Not_Significative = is_significative[["FALSE"]], Up_Regulated = direction[["up"]], Down_Regulated = direction[["down"]] ) utils::write.table( x = sig, file = base::as.character(x = snakemake@output[["qc"]]), sep = "\t" ) base::message("QC table saved") } if ("csaw" %in% base::names(snakemake@output)) { base::saveRDS( object = counts, file = base::as.character(x = snakemake@output[["csaw"]]) ) base::message("csaw results saved") } if ("edger" %in% base::names(snakemake@ouptput)) { base::saveRDS( object = results, file = base::as.character(x = snakemake@output[["edger"]]) ) base::message("edgeR results saved") } base::message("Process over") # Proper syntax to close the connection for the log file # but could be optional for Snakemake wrapper base::sink(type = "message") base::sink() |
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 196 197 198 199 200 201 202 203 204 205 206 207 208 209 210 211 212 213 214 215 | log_file <- file(snakemake@log[[1]], open = "wt") sink(log_file) sink(log_file, type = "message") # Load libraries base::library(package = "csaw", character.only = TRUE) base::message("Libraries loaded") counts <- base::readRDS( file = base::as.character(x = snakemake@input[["counts"]]) ) log_threshold <- 1.1 if ("log_threshold" %in% base::names(snakemake@params)) { log_threshold <- as.numeric(snakemake@params[["log_threshold"]]) } filter_method <- "average_log_cpm" if ("filter_method" %in% base::names(x = snakemake@params)) { filter_method <- base::as.character( x = snakemake@params[["filter_method"]] ) } keep <- base::data.frame( "Kept" = 0, "Filtered" = 0, "PercentFiltered" = 0, "PercentKept" = 0 ) base::message("Input data loaded") # Filtering counts if (filter_method == "average_log_cpm") { # Filter based on average CPM counts. # not that good in case of large sequencing libraries # not that good in case of large variability in sequencing library sizes base::message("Filtering based on count size (aveLogCPM)") min_cpm <- 5 if ("min_cpm" %in% base::names(snakemake@params)) { min_cpm <- base::as.numeric(x = snakemake@params[["min_cpm"]]) } abundance <- edgeR::aveLogCPM(SummarizedExperiment::asDGEList(counts)) keep <- abundances > aveLogCPM(min_cpm, lib.size = mean(counts$totals)) counts <- counts[keep, ] } else if (filter_method == "proportion") { # Filter based on estimated proportion of signal of interest / noise # Not that good if site proportion varies across samples base::message("Filtering based on signal / noise proportion") proportion <- 0.999 if ("proportion" %in% base::names(x = snakemake@params)) { proportion <- base::as.numeric(x = snakemake@params[["proportion"]]) } keep <- csaw::filterWindowsProportion(counts)$filter > proportion counts <- counts[keep, ] } else if (filter_method == "global_enrichment") { # Filtering by global enrichment. The degree of background enrichment # is estimated by counting reads into large bins across the genome. # Not that good if there are differences in the background coverage # across the genome. base::message("Filtering based on global enrichment") binned <- base::readRDS( file = base::as.character(x = snakemake@input[["binned"]]) ) filter_stat <- csaw::filterWindowsGlobal( data = counts, background = binned ) keep <- filter_stat$filter > log2(log_threshold) counts <- counts[keep, ] } else if (filter_method == "local_enrichment") { # Mimicking single-sample peak callers. # Not good if any peaks are in the neighborhood ! Please make # sure there are not peak clusters of any kind. base::message("Filtering based on local enrichment") neighbor <- base::readRDS( file = base::as.character(x = snakemake@input[["neighbor"]]) ) filter_stat <- csaw::filterWindowsLocal( data = counts, background = neighbor ) keep <- filter_stat$filter > log2(log_threshold) counts <- counts[keep, ] } else if (filter_method == "local_maxima") { # Use highest average abundance within 1kbp on either side. # Very aggressive, use only in datasets with sharp binding. base::message("Filtering based on local peak maxima") maxed <- base::readRDS( file = base::as.character(x = snakemake@input[["maxed"]]) ) filter_stat <- csaw::filterWindowsLocal( data = counts, background = maxed ) keep <- filter_stat$filter > log2(log_threshold) counts <- counts[keep, ] } else if (filter_method == "input_controls") { # Use negative controls for ChIP-seq to account for # both sequencing and mapping biases in ChIP-seq data base::message("Filtering based on negative controls (aka Input)") input_counts <- base::readRDS( file = base::as.character(x = snakemake@input[["input_counts"]]) ) base::message("Input counts loaded") counts_binned <- base::readRDS( file = base::as.character(x = snakemake@input[["binned"]]) ) base::message("Tested (binned) counts loaded") input_binned <- base::readRDS( file = base::as.character(x = snakemake@input[["input_binned"]]) ) base::message("Input (binned) counts loaded") scale_info <- csaw::scaleControlFilter( data.bin = counts_binned, back.bin = input_binned ) base::message("Scale control filter acquired") filter_stat <- csaw::filterWindowsControl( data = counts, background = input_counts, prior.count = 2, scale.info = scale_info ) base::message("Negative control signal filtered") keep <- filter_stat$filter > log2(log_threshold) counts <- counts[keep, ] } else { base::stop("Unknown filter method") } k <- base::summary(object = keep) base::message("Number of targets passing filters") base::print(k) keep <- base::as.data.frame(keep) keep$Kept <- k[["TRUE"]] keep$Filtered <- k[["FALSE"]] keep$PercentFiltered <- keep$Filtered / (keep$Kept + keep$Filtered) keep$PercentKept <- keep$Kept / (keep$Kept + keep$Filtered) # Saving results utils::write.table( x = keep, file = base::as.character(x = snakemake@output[["qc"]]), sep = "\t" ) base::message("QC table saved") if ("png" %in% base::names(x = snakemake@output)) { # Saving QC plot grDevices::png( filename = base::as.character(x = snakemake@output[["png"]]), width = 1024, height = 768 ) graphics::hist( filter_stat$filter, main = "", breaks = 50, xlab = "Background abundance (log2-CPM)" ) graphics::abline(v = log2(log_threshold), col = "red") grDevices::dev.off() base::message("QC plot saved") } base::saveRDS( object = counts, file = base::as.character(x = snakemake@output[["rds"]]) ) base::message("RDS saved, process over") # Proper syntax to close the connection for the log file # but could be optional for Snakemake wrapper base::sink(type = "message") base::sink() |
8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 | base::library(package = "BiocParallel", character.only = TRUE) base::library(package = "csaw", character.only = TRUE) base::message("Library loaded") # Loading input files read_params <- base::readRDS( file = base::as.character(x = snakemake@input[["read_params"]]) ) read_params <- csaw::reform(x = read_params, dedup = TRUE) base::message("Read parameters loaded, with dedup set to TRUE") design <- base::readRDS(file = snakemake@input[["design"]]) base::message("Experimental design loaded with BamFiles") max_delay <- 800 if (snakemake@threads > 1) { BiocParallel::register( BPPARAM = BiocParallel::MulticoreParam(workers = snakemake@threads) ) options("mc.cores" = snakemake@threads) base::message("Process multi-threaded") } # Compute fragment size with cross-correlation correlation <- csaw::correlateReads( bam.files = design$BamPath, max.dist = max_delay, cross = TRUE, param = read_params ) frag_length <- csaw::maximizeCcf(correlation, ignore = 100) base::message("Fragment length computed") base::saveRDS( object = frag_length, file = base::as.character(x = snakemake@output[["fragment_length"]]) ) base::message("Fragment length saved") grDevices::png( filename = base::as.character(x = snakemake@output[["png"]]), width = 1024, height = 788 ) graphics::plot( x = 0:max_delay, y = correlation, xlab = "Delay (bp)", ylab = "CCF", type = "l" ) graphics::abline(v = frag_length, col = "red") graphics::text( paste(frag_length, "bp"), x = frag_length, y = min(correlation), pos = "4", col = "red" ) grDevices::dev.off() base::message("Plot saved, process over") # Proper syntax to close the connection for the log file # but could be optional for Snakemake wrapper base::sink(type = "message") base::sink() |
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 | log_file <- file(snakemake@log[[1]], open = "wt") sink(log_file) sink(log_file, type = "message") # Load libraries base::library(package = "csaw", character.only = TRUE) base::library(package = "edgeR", character.only = TRUE) base::message("Libraries loaded") norm_method <- "composition" if ("norm_method" %in% base::names(snakemake@params)) { norm_method <- base::as.character(x = snakemake@params[["norm_method"]]) } design <- base::readRDS( file = base::as.character(x = snakemake@input[["design"]]) ) base::message("Design loaded") counts <- base::readRDS( file = base::as.character(x = snakemake@input[["counts"]]) ) base::message("Counts loaded") if (norm_method == "composition") { # Highly enriched regions consume more sequencing resources and # thereby suppress the representation of other regions. Differences # in the magnitude of suppression between libraries can lead to # spurious DB calls. Scaling by library size fails to correct for this as # composition biases can still occur in libraries of the same size. base::message("Normalizing composition bias") binned <- base::readRDS( file = base::as.character(x = snakemake@input[["bins"]]) ) counts <- csaw::normFactors( object = binned, se.out = counts ) adj_counts <- edgeR::cpm( y = csaw::asDGEList(object = binned, log = TRUE) ) normfacs <- counts$norm.factors } else if (norm_method == "efficiency") { # Efficiency biases in ChIP-seq data refer to fold changes in enrichment # that are introduced by variability in IP efficiencies between libraries. base::message("Normalizing efficiency bias") counts <- csaw::normFactors( object = counts, se.out = TRUE ) adj_counts <- edgeR::cpm( y = csaw::asDGEList(object = counts, log = TRUE) ) normfacs <- counts$norm.factors } base::saveRDS( object = adj_counts, file = base::as.character(x = snakemake@output["adj_counts"]) ) base::message("Adjusted counts saved") base::saveRDS( object = counts, file = base::as.character(x = snakemake@output[["rds"]]) ) base::message("RDS saved, process over") # Proper syntax to close the connection for the log file # but could be optional for Snakemake wrapper base::sink(type = "message") base::sink() |
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 | log_file <- file(snakemake@log[[1]], open = "wt") sink(log_file) sink(log_file, type = "message") # Load libraries base::library(package = "csaw", character.only = TRUE) base::library(package = "rtracklayer", character.only = TRUE) base::message("Libraries loaded") standard_chrom <- c(1:23, "X", "Y") if ("organism" %in% base::names(x = snakemake@params)) { if (base::as.character(x = snakemake@params[["organism"]]) == "mm10") { standard_chrom <- c(1:19, "X", "Y") } } blacklist <- base::as.character(x = snakemake@input[["blacklist"]]) blacklist <- rtracklayer::import(blacklist) # Build command line extra <- "minq=20, restrict=standard_chrom, discard=blacklist" if ("extra" %in% base::names(x = snakemake@params)) { extra <- base::as.character(x = snakemake@params[["extra"]]) } command <- base::paste0( "csaw::readParam(", extra, ")" ) base::message("Command line:") base::print(command) # Run command line read_params <- base::eval(base::parse(text = command)) # Save results base::saveRDS( object = read_params, file = snakemake@output[["rds"]] ) base::message("RDS saved, process over") # Proper syntax to close the connection for the log file # but could be optional for Snakemake wrapper base::sink(type = "message") base::sink() |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 | base::message("Loading libraries ... ") suppressPackageStartupMessages(library("ATACseqQC")) suppressPackageStartupMessages(library("TxDb.Hsapiens.UCSC.hg38.knownGene")) suppressPackageStartupMessages(library("BSgenome.Hsapiens.UCSC.hg38")) suppressPackageStartupMessages(library("phastCons100way.UCSC.hg38")) suppressPackageStartupMessages(library("MotifDb")) suppressPackageStartupMessages(library("ChIPpeakAnno")) suppressPackageStartupMessages(library("Rsamtools")) base::message("Libraries loaded.") # base::message("Setting sequence level style...") # seqlevelsStyle(TxDb.Hsapiens.UCSC.hg38.knownGene) <- "Ensembl" # seqlevelsStyle(BSgenome.Hsapiens.UCSC.hg38) <- "Ensembl" # base::message("Database chromosome renamed.") base::message("Acquiering bam file...") bamfile <- BamFile( file = base::as.character(x = snakemake@input[["bam"]]) ) name <- base::as.character(x = snakemake@params[["name"]]) base::print(bamfile) base::print(name) base::message("BamFiles identified") base::message("Reading bam tags...") possibleTag <- list( "integer" = c( "AM", "AS", "CM", "CP", "FI", "H0", "H1", "H2", "HI", "IH", "MQ", "NH", "NM", "OP", "PQ", "SM", "TC", "UQ" ), "character" = c( "BC", "BQ", "BZ", "CB", "CC", "CO", "CQ", "CR", "CS", "CT", "CY", "E2", "FS", "LB", "MC", "MD", "MI", "OA", "OC", "OQ", "OX", "PG", "PT", "PU", "Q2", "QT", "QX", "R2", "RG", "RX", "SA", "TS", "U2" ) ) bamTop100 <- scanBam( BamFile(bamfile$path, yieldSize = 100), param = ScanBamParam(tag=unlist(possibleTag)) )[[1]]$tag tags <- names(bamTop100)[lengths(bamTop100) > 0] base::print(tags) base::message("Tags Acquired") base::message("Retrieving sequence level informations...") seqlev <- as.vector( sapply(c(1:22, "X", "Y"), function(chrom) paste0("chr", chrom)) ) seqinformation <- seqinfo(TxDb.Hsapiens.UCSC.hg38.knownGene) which <- as(seqinformation[seqlev], "GRanges") base::print(which) base::message("Sequences retrived") base::message("Loading bam file...") bamdata <- readBamFile( bamFile = bamfile$path, bigFile = TRUE, asMates = TRUE, tags = tags, which = which, ) base::message("Bam file loaded") base::message("Shifting bam...") shiftedBamfile <- base::as.character(x = snakemake@output[["shifted"]]) shiftedBamdir <- base::dirname(shiftedBamfile) print(shiftedBamdir) base::dir.create( path = shiftedBamdir, recursive = TRUE ) bamdata <- shiftGAlignmentsList( gal = bamdata, outbam = shiftedBamfile ) print(bamdata) base::message("Shift OK") base::message("Acquiering motif...") motif_name <- base::as.character(x = snakemake@params[["motif"]]) motif <- query(MotifDb, c(motif_name)) motif <- as.list(motif) print(motif[[1]], digits = 2) base::message("Motif retrieved.") base::message("plot Footprints...") genome <- Hsapiens print(genome) png( filename = snakemake@output[["png"]], width = 1024, height = 768, units = "px" ) sigs <- factorFootprints( shiftedBamfile, pfm = motif[[1]], genome = genome, min.score = "90%", seqlev = c(1:22, "X", "Y"), upstream = 100, downstream = 100 ) dev.off() base::message("Done.") base::save.image(file = base::as.character(x = snakemake@output[["rda"]])) base::message("Process over") |
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 | log_file <- file(snakemake@log[[1]], open = "wt") sink(log_file) sink(log_file, type = "message") # Load libraries base::library(package = "MEDIPS", character.only = TRUE) base::library(package = "BSgenome.Hsapiens.UCSC.hg38", character.only = TRUE) base::library(package = "BSgenome.Mmusculus.UCSC.mm10", character.only = TRUE) results <- base::readRDS( file = base::as.character(x = snakemake@input[["meth"]]) ) base::message("Dataset loaded") # Build command line extra <- "results = results" if ("extra" %in% base::names(x = snakemake@params)) { extra <- base::paste( extra, base::as.character(x = snakemake@params[["extra"]]), sep = ", " ) } command <- base::paste0( "MEDIPS.selectSig(", extra, ")" ) base::message("Command line:") base::paste(command) # Launch analysis edger_results <- base::eval(base::parse(text = command)) base::message("Differential peak coverage perfomed") # Optionally merge close peaks if ("merge_distance" %in% base::names(x = snakemake@params)) { edger_results <- MEDIPS.mergeFrames( frames = edger_results, distance = base::as.numeric(x = snakemake@params[["merge_distance"]]) ) base::message("Close peaks merged") } # Save results base::saveRDS( object = edger_results, file = base::as.character(x = snakemake@output[["rds"]]) ) utils::write.table( x = edger_results, file = base::as.character(x = snakemake@output[["tv"]]), sep = "\t", quote = FALSE ) base::message("Process over") |
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 | log_file <- file(snakemake@log[[1]], open = "wt") sink(log_file) sink(log_file, type = "message") # Load libraries base::library(package = "MEDIPS", character.only = TRUE) base::library(package = "BSgenome.Hsapiens.UCSC.hg38", character.only = TRUE) base::library(package = "BSgenome.Mmusculus.UCSC.mm10", character.only = TRUE) base::library(package = "DNAcopy", character.only = TRUE) base::message("Libraries loaded") mset1 <- base::readRDS(file = base::as.character(x = "mset1")) extra <- "MSet1 = mset1, diff.method = 'edgeR', p.adj = 'bonferroni'" base::message("MSet1 loaded") mset2 <- NULL if ("mset2" %in% base::names(snakemake@input)) { mset2 <- base::readRDS(file = base::as.character(x = "mset2")) extra <- base::paste(extra, "MSet2 = mset2", sep = ", ") base::message("MSet2 loaded") } cset <- NULL if ("cset" %in% base::names(snakemake@input)) { cset <- base::readRDS(file = base::as.character(x = "cset")) extra <- base::paste(extra, "CSet = cset", sep = ", ") base::message("CSet loaded") } iset1 <- NULL if ("iset1" %in% base::names(snakemake@input)) { iset1 <- iset1base::readRDS(file = base::as.character(x = "iset1")) extra <- base::paste(extra, "ISet1 = iset1", sep = ", ") base::message("ISet1 loaded") } iset2 <- NULL if ("iset2" %in% base::names(snakemake@input)) { iset2 <- iset1base::readRDS(file = base::as.character(x = "iset2")) extra <- base::paste(extra, "ISet2 = iset2", sep = ", ") base::message("ISet2 loaded") } if (! (base::is.null(iset1) && base::is.null(iset2))) { extra <- base::paste(extra, "CNV = TRUE", sep = ", ") base::message( "copy number variation will be tested by ", "applying the package DNAcopy to the ", "window-wise log-ratios calculated based on ", "the the means per group" ) } if (! (base::is.null(mset2) && base::is.null(cset))) { extra <- base::paste(extra, "MeDIP = TRUE", sep = ", ") base::message( "CpG density dependent relative methylation scores ", "(rms) will be calculated for the MEDIPS SETs" ) } medip_meth_command <- base::paste0( "MEDIPS::MEDIPS.meth(", extra, ")" ) base::message("Command line used to determine coverage and analysis methods:") base::message(medip_meth_command) coverage_profiles <- base::eval(base::parse(text = medip_meth_command)) base::saveRDS( object = coverage_profiles, file = base::as.character(x = snakemake@output[["rds"]]) ) # Proper syntax to close the connection for the log file # but could be optional for Snakemake wrapper sink(type = "message") sink() |
16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 | __author__ = "Thibault Dayris" __copyright__ = "Copyright 2019, Thibault Dayris" __email__ = "[email protected]" __license__ = "MIT" import os import os.path as op import logging from random import randint from snakemake.shell import shell from tempfile import TemporaryDirectory # Logging behaviour logging.basicConfig(filename=snakemake.log[0], filemode="w", level=logging.DEBUG) # Prepare logging log = snakemake.log_fmt_shell(stdout=False, stderr=True, append=True) extra_cp = snakemake.params.get("extra", "--force --recursive --verbose") extra_iget = snakemake.params.get("extra_irods", "-vKrf") extra_ln = snakemake.params.get("extra_ln", "--relative --verbose --symbolic --force") extra_rsync = snakemake.params.get( "extra_rsync", "--verbose --recursive --checksum --human-readable --partial --progress", ) use_cp_over_rsync = snakemake.params.get("use_cp_over_rsync", False) master_node = snakemake.params.get("master_node", "flamingo-lg-01") parallel = snakemake.params.get("internal_parallel", False) logging.info("Extra acquired") if ("-N" not in extra_iget) and snakemake.threads > 1: max_threads = snakemake.threads if os.uname()[1] == master_node and max_threads > 4: logging.warning( "More than 4 threads per copy on master node is not allowed. " "Max threads set to 4. This does not change your original " "threading reservation. Run this script on a computing node, " "or lower the number of threads." ) max_threads = 4 if parallel: logging.info("Internal parallelization") max_threads = 1 extra_iget += f" -N {max_threads} " # No node can access a cold storage # these files must be copied. However, # any file else where should be symlinked! cold_storage = ("/mnt/isilon", "/mnt/archivage", "/mnt/install") if "cold_storage" in snakemake.params.keys(): cold_storage = snakemake.params["cold_storage"] logging.info("Mounting point qualified") def cat_files(dest: str, *src: str, log: str = log) -> None: command = f"cat {' '.join(src)} > {dest} {log}" logging.info(command) shell(command) def bash_copy( src: str, dest: str, extra_cp: str = extra_cp, extra_rsync: str = extra_rsync, extra_ln: str = extra_ln, log: str = log, cold: str = cold_storage, use_cp: bool = use_cp_over_rsync, ) -> None: if not src.startswith(cold): logging.info("Operation on cold storage") command = f"ln {extra_ln} {src} {dest} {log}" logging.info(command) shell(command) else: logging.info("Operation on active storage") command = "cp" if use_cp else "rsync" extra = extra_cp if use_cp else extra_rsync command = f"{command} {extra} {src} {dest} {log}" logging.info(command) shell(command) def iRODS_copy(src: str, dest: str, extra: str = extra_iget, log: str = log) -> None: command = f"iget {extra} {src} {dest} {log}" logging.info(command) shell(command) def copy(src: str, dest: str) -> None: if src.startswith("/odin/kdi/dataset/"): logging.info("Using iRODS") iRODS_copy(src, dest) else: logging.info("*Not* using iRODS") bash_copy(src, dest) def copy_then_concat(dest: str, *src: str) -> None: logging.info("Performing conatenation") with TemporaryDirectory() as tmpdir: outfiles = [] for path in src: tmp_dest = f"{tmpdir}/{os.path.basename(path)}.{randint(0, 100_000_000)}" copy(path, tmp_dest) outfiles.append(tmp_dest) cat_files(dest, *outfiles) def copy_or_concat(src: str, dest: str) -> None: if "," in src: copy_then_concat(dest, *src.split(",")) else: copy(src, dest) output_directory = op.realpath(op.dirname(snakemake.output[0])) logging.debug(output_directory) sources = snakemake.params.get("input", snakemake.input) logging.debug(sources) destinations = snakemake.output logging.debug(destinations) if len(destinations) == 1: # Then there is only one directory as a destination destination = op.realpath(str(destinations)) command = f"mkdir --parents --verbose {op.dirname(destination)} {log}" logging.info(command) shell(command) if isinstance(sources, str): copy_or_concat(sources, destination) else: for source in sources: copy_or_concat(source, destination) elif len(sources) == len(destinations): # Then there must be as many paths as source for source, destination in zip(sources, destinations): command = f"mkdir --parents --verbose {op.dirname(destination)} {log}" logging.info(command) shell(command) copy_or_concat(source, destination) else: logging.error(f"nb of input: {len(sources)}, nb of output: {len(destinations)}") logging.debug(sources) logging.debug(destinations) raise ValueError("Number of input and output paths do not match.") |
8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 | base::library(package = "Rsamtools", character.only = TRUE) base::message("Library loaded") bam_file <- Rsamtools::BamFile( file = base::as.character(x = snakemake@input[["bam"]]), index = base::as.character(x = snakemake@input[["bai"]]) ) base::message("Bam file created") base::saveRDS( object = bam_file, file = base::as.character(x = snakemake@output["rds"]) ) base::message("Process over") # Proper syntax to close the connection for the log file # but could be optional for Snakemake wrapper base::sink(type = "message") base::sink() |
9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 | base::library(package = "Rsamtools", character.only = TRUE) base::message("Library loaded") # Loading input data rds_paths <- base::sapply( snakemake@input[["bam_files"]], function(rds_path) base::readRDS(file = base::as.character(x = rds_path)) ) base::message("BamFiles loaded") design <- utils::read.table( file = base::as.character(x = snakemake@input[["design"]]), header = TRUE, sep = "\t", stringsAsFactors = FALSE ) base::rownames(design) <- design$Sample_id base::message("Design loaded") # Building quality control table design$BamPath <- rds_paths base::message("Design enhanced") diagnostics <- base::list() for (i in base::seq_along(design$BamPath)) { bam <- design$BamPath[[i]] total_reads <- Rsamtools::countBam(file = bam)$records params <- Rsamtools::ScanBamParam( flag = Rsamtools::scanBamFlag(isUnmapped = FALSE) ) tota_mapped <- Rsamtools::countBam(file = bam, param = params) params <- Rsamtools::ScanBamParam( flag = Rsamtools::scanBamFlag(isUnmapped = FALSE, isDuplicate = TRUE) ) marked <- Rsamtools::countBam(bam, param = params)$records diagnostics[[i]] <- c( Total = total_reads, Mapped = tota_mapped, Marked = marked ) } base::print(diagnostics) diag_stats <- base::data.frame(base::do.call(rbind, diagnostics)) base::rownames(diag_stats) <- design$Sample_id diag_stats$Prop.mapped <- ( as.numeric( x = diag_stats[["Mapped.records"]] ) / as.numeric( x = diag_stats[["Total"]] ) ) * 100 diag_stats$Prop.marked <- ( as.numeric( x = diag_stats[["Marked"]] ) / as.numeric( x = diag_stats[["Mapped.records"]] ) ) * 100 base::message("Quality controls performed") # diag_stats <- data.frame(t(sapply(diag_stats, c))) diag_stats <- data.frame(lapply(diag_stats, as.character), stringsAsFactors=FALSE) qc_path <- base::as.character(x = snakemake@output[['qc']]) print(diag_stats) print(qc_path) print(str(diag_stats)) # Saving results utils::write.table( x = diag_stats, file = qc_path, sep = "\t", row.names = TRUE, col.names = TRUE ) base::message("QC saved") base::saveRDS( object = design, file = base::as.character(x = snakemake@output[["design"]]) ) base::message("Design saved") # Proper syntax to close the connection for the log file # but could be optional for Snakemake wrapper base::sink(type = "message") base::sink() |
Support
- Share:
-

-

-

- Future updates
Related Workflows





