Jupyter Notebook Mutation Free Energy Calculations using BioExcel Building Blocks (biobb)



Help improve this workflow!
This workflow has been published but could be further improved with some additional meta data:- Keyword(s) in categories input, output
You can help improve this workflow by suggesting the addition or removal of keywords, suggest changes and report issues, or request to become a maintainer of the Workflow .
Mutation Free Energy Calculations using BioExcel Building Blocks (biobb)
Based on the official pmx tutorial .
This tutorial aims to illustrate how to compute a fast-growth mutation free energy calculation, step by step, using the BioExcel Building Blocks library (biobb) . The particular example used is the Staphylococcal nuclease protein (PDB code 1STN), a small, minimal protein, appropriate for a short tutorial.
The non-equilibrium free energy calculation protocol performs a fast alchemical transition in the direction WT->Mut and back Mut->WT . The two equilibrium trajectories needed for the tutorial, one for Wild Type (WT) and another for the Mutated (Mut) protein (Isoleucine 10 to Alanine -I10A-), have already been generated and are included in this example. We will name WT as stateA and Mut as stateB .
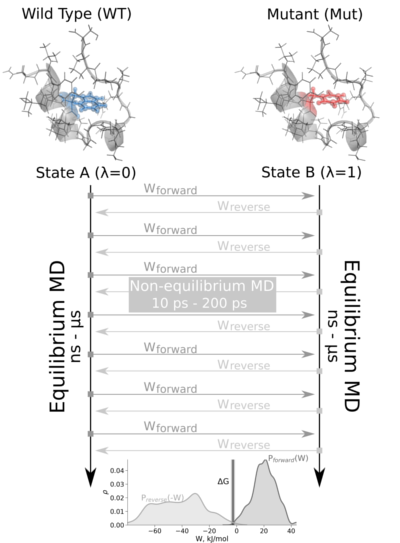
The tutorial calculates the free energy difference in the folded state of a protein. Starting from two 1ns-length independent equilibrium simulations (WT and mutant), snapshots are selected to start fast (50ps) transitions driving the system in the forward (WT to mutant) and reverse (mutant to WT) directions, and the work values required to perform these transitions are collected. With these values, Crooks Gaussian Intersection (CGI), Bennett Acceptance Ratio (BAR) and Jarzynski estimator methods are used to calculate the free energy difference between the two states.
![]()
Code Snippets
2 3 4 5 6 7 8 9 10 11 12 | import os import zipfile cwd = os.getcwd() gmxlib = os.getenv('CONDA_PREFIX')+'/lib/python3.10/site-packages/pmx/data/mutff/' stateA_traj = cwd + "/pmx_tutorial/stateA_1ns.xtc" stateA_tpr = cwd + "/pmx_tutorial/stateA.tpr" stateB_traj = cwd + "/pmx_tutorial/stateB_1ns.xtc" stateB_tpr = cwd + "/pmx_tutorial/stateB.tpr" |
16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 | # GMXTrjConvStrEns: extract an ensemble of snapshots from a GROMACS trajectory file # Import module from biobb_analysis.gromacs.gmx_trjconv_str_ens import gmx_trjconv_str_ens #### State A #### # Create prop dict and inputs/outputs (StateA) output_framesA = 'stateA_frames.zip' prop = { 'selection' : 'System', 'start': 1, # To be changed to generate as many snapshots as needed 'end': 1000, # To be changed to generate as many snapshots as needed 'dt': 200, # To be changed to generate as many snapshots as needed 'output_name': 'frameA', 'output_type': 'pdb' } # Create and launch bb (StateA) gmx_trjconv_str_ens(input_traj_path=stateA_traj, input_top_path=stateA_tpr, output_str_ens_path=output_framesA, properties=prop) # Extract stateA (WT) frames with zipfile.ZipFile(output_framesA, 'r') as zip_f: zip_f.extractall() stateA_pdb_list = zip_f.namelist() #### State B #### # Create prop dict and inputs/outputs (StateB) output_framesB = 'stateB_frames.zip' prop = { 'selection' : 'System', 'start': 1, # To be changed to generate as many snapshots as needed 'end': 1000, # To be changed to generate as many snapshots as needed 'dt': 200, # To be changed to generate as many snapshots as needed 'output_name': 'frameB', 'output_type': 'pdb' } # Create and launch bb (StateB) gmx_trjconv_str_ens(input_traj_path=stateB_traj, input_top_path=stateB_tpr, output_str_ens_path=output_framesB, properties=prop) # # Extract stateB (Mutant) frames with zipfile.ZipFile(output_framesB, 'r') as zip_f: zip_f.extractall() stateB_pdb_list = zip_f.namelist() |
72 73 74 75 | # Prepare Mutation Free Energy calculation for ONE PARTICULAR frame of each state # (to be repeated for the rest of the frames) pdbA = stateA_pdb_list[0] pdbB = stateB_pdb_list[0] |
79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 | # pmx mutate: Mutate command from pmx package # Import module from biobb_pmx.pmxbiobb.pmxmutate import pmxmutate #### State A (WT->Mut) #### # Create prop dict and inputs/outputs output_structure_mutA = 'mutA.pdb' prop = { 'force_field' : 'amber99sb-star-ildn-mut', 'mutation_list' : '10Ala', 'binary_path' : 'pmx', 'gmx_lib' : gmxlib } # Create and launch bb pmxmutate(input_structure_path=pdbA, output_structure_path=output_structure_mutA, properties=prop) #### State B (Mut->WT) #### # Create prop dict and inputs/outputs output_structure_mutB = 'mutB.pdb' prop = { 'force_field' : 'amber99sb-star-ildn-mut', 'mutation_list' : '10Ile', 'binary_path' : 'pmx', 'gmx_lib' : gmxlib } # Create and launch bb pmxmutate(input_structure_path=pdbB, output_structure_path=output_structure_mutB, properties=prop) |
118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 | # Create system topology # Import module from biobb_gromacs.gromacs.pdb2gmx import pdb2gmx #### State A (WT->Mut) #### # Create inputs/outputs output_pdb2gmxA_gro = 'pdb2gmxA.gro' output_pdb2gmxA_top_zip = 'pdb2gmxA_top.zip' prop = { 'force_field' : 'amber99sb-star-ildn-mut', 'gmx_lib' : gmxlib } # Create and launch bb pdb2gmx(input_pdb_path=output_structure_mutA, output_gro_path=output_pdb2gmxA_gro, output_top_zip_path=output_pdb2gmxA_top_zip, properties=prop) #### State B (Mut->WT) #### # Create inputs/outputs output_pdb2gmxB_gro = 'pdb2gmxB.gro' output_pdb2gmxB_top_zip = 'pdb2gmxB_top.zip' prop = { 'force_field' : 'amber99sb-star-ildn-mut', 'gmx_lib' : gmxlib } # Create and launch bb pdb2gmx(input_pdb_path=output_structure_mutB, output_gro_path=output_pdb2gmxB_gro, output_top_zip_path=output_pdb2gmxB_top_zip, properties=prop) |
159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 196 197 198 | # pmx gentop: Gentop command (Generate Hybrid Topology) from pmx package # Import module from biobb_pmx.pmxbiobb.pmxgentop import pmxgentop #### State A (WT->Mut) #### # Create prop dict and inputs/outputs output_pmxtopA_top_zip = 'pmxA_top.zip' output_pmxtopA_log = 'pmxA_top.log' prop = { 'force_field' : 'amber99sb-star-ildn-mut', 'binary_path' : 'pmx', 'gmx_lib' : gmxlib } #Create and launch bb pmxgentop(input_top_zip_path=output_pdb2gmxA_top_zip, output_top_zip_path=output_pmxtopA_top_zip, output_log_path=output_pmxtopA_log, properties=prop) #### State B (Mut->WT) #### # Create prop dict and inputs/outputs output_pmxtopB_top_zip = 'pmxB_top.zip' output_pmxtopB_log = 'pmxB_top.log' prop = { 'force_field' : 'amber99sb-star-ildn-mut', 'binary_path' : 'pmx', 'gmx_lib' : gmxlib } # Create and launch bb pmxgentop(input_top_zip_path=output_pdb2gmxB_top_zip, output_top_zip_path=output_pmxtopB_top_zip, output_log_path=output_pmxtopB_log, properties=prop) |
202 203 204 205 206 207 208 209 210 211 212 213 214 215 216 217 218 | # Gromacs make_ndx: GROMACS Make index command from biobb_gromacs package # IMPORTANT: Only needed for stateB # Import module from biobb_gromacs.gromacs.make_ndx import make_ndx # Create prop dict and inputs/outputs output_ndx = 'indexB.ndx' prop = { 'selection' : 'a D*\n0 & ! 19\nname 20 FREEZE' } # Create and launch bb make_ndx(input_structure_path=output_pdb2gmxB_gro, output_ndx_path=output_ndx, properties=prop) |
222 223 224 225 226 227 228 229 230 231 232 233 234 235 236 237 238 239 240 241 242 243 244 245 246 247 248 249 250 | # Grompp: Creating portable binary run file for dummy atoms energy minimization from biobb_gromacs.gromacs.grompp import grompp #### State B (Mut->WT) #### # Create prop dict and inputs/outputs output_tpr_min = 'em.tpr' prop = { 'gmx_lib' : gmxlib, 'mdp':{ 'integrator' : 'steep', 'emtol': '100', 'dt': '0.001', 'nsteps':'10000', 'nstcomm': '1', 'nstcalcenergy': '1', 'freezegrps' : 'FREEZE', 'freezedim' : "Y Y Y" }, 'simulation_type': 'minimization' } # Create and launch bb grompp(input_gro_path=output_pdb2gmxB_gro, input_top_zip_path=output_pmxtopB_top_zip, input_ndx_path=output_ndx, output_tpr_path=output_tpr_min, properties=prop) |
254 255 256 257 258 259 260 261 262 263 264 265 266 267 268 | # Mdrun: Running minimization from biobb_gromacs.gromacs.mdrun import mdrun # Create prop dict and inputs/outputs output_min_trr = 'emout.trr' output_min_gro = 'emout.gro' output_min_edr = 'emout.edr' output_min_log = 'emout.log' # Create and launch bb mdrun(input_tpr_path=output_tpr_min, output_trr_path=output_min_trr, output_gro_path=output_min_gro, output_edr_path=output_min_edr, output_log_path=output_min_log) |
272 273 274 275 276 277 278 279 280 281 282 283 284 | # GMXEnergy: Getting system energy by time from biobb_analysis.gromacs.gmx_energy import gmx_energy # Create prop dict and inputs/outputs output_min_ene_xvg = 'min_ene.xvg' prop = { 'terms': ["Potential"] } # Create and launch bb gmx_energy(input_energy_path=output_min_edr, output_xvg_path=output_min_ene_xvg, properties=prop) |
288 289 290 291 292 293 294 295 296 297 298 299 300 301 302 303 304 305 306 307 308 309 310 311 312 313 | import plotly import plotly.graph_objs as go # Read data from file and filter energy values higher than 1000 Kj/mol^-1 with open(output_min_ene_xvg,'r') as energy_file: x,y = map( list, zip(*[ (float(line.split()[0]),float(line.split()[1])) for line in energy_file if not line.startswith(("#","@")) if float(line.split()[1]) < 1000 ]) ) plotly.offline.init_notebook_mode(connected=True) fig = ({ "data": [go.Scatter(x=x, y=y)], "layout": go.Layout(title="Energy Minimization", xaxis=dict(title = "Energy Minimization Step"), yaxis=dict(title = "Potential Energy KJ/mol-1") ) }) plotly.offline.iplot(fig) |
317 318 319 320 321 322 323 324 325 326 327 328 329 330 331 332 333 334 335 336 337 338 339 340 341 342 343 344 345 346 347 348 349 350 351 352 353 354 355 356 357 358 359 360 361 362 363 | # Grompp: Creating portable binary run file for system equilibration from biobb_gromacs.gromacs.grompp import grompp #### State A (WT->Mut) #### # Create prop dict and inputs/outputs output_tprA_eq = 'eqA_20ps.tpr' prop = { 'gmx_lib' : gmxlib, 'mdp':{ 'nsteps':'10000', 'nstcomm' : '1', 'dt':'0.001', 'nstcalcenergy' : '1' }, 'simulation_type': 'free' } #Create and launch bb grompp(input_gro_path=output_pdb2gmxA_gro, input_top_zip_path=output_pmxtopA_top_zip, output_tpr_path=output_tprA_eq, properties=prop) #### State B (Mut->WT) #### # Create prop dict and inputs/outputs output_tprB_eq = 'eqB_20ps.tpr' prop = { 'gmx_lib' : gmxlib, 'mdp':{ 'nsteps':'10000', # 10000 steps x 1fs (timestep) = 10ps 'dt':'0.001', # 1 fs of timestep, to properly equilibrate dummy atoms 'nstcomm' : '1', 'nstcalcenergy' : '1' }, 'simulation_type': 'free' } #Create and launch bb grompp(input_gro_path=output_min_gro, input_top_zip_path=output_pmxtopB_top_zip, output_tpr_path=output_tprB_eq, properties=prop) |
367 368 369 370 371 372 373 374 375 376 377 378 379 380 381 382 383 384 385 386 387 388 389 390 391 392 393 394 395 396 397 398 | # Mdrun: Running equilibration from biobb_gromacs.gromacs.mdrun import mdrun #### State A (WT->Mut) #### # Create prop dict and inputs/outputs output_eqA_trr = 'eqoutA.trr' output_eqA_gro = 'eqoutA.gro' output_eqA_edr = 'eqoutA.edr' output_eqA_log = 'eqoutA.log' # Create and launch bb mdrun(input_tpr_path=output_tprA_eq, output_trr_path=output_eqA_trr, output_gro_path=output_eqA_gro, output_edr_path=output_eqA_edr, output_log_path=output_eqA_log) #### State B (Mut->WT) #### # Create prop dict and inputs/outputs output_eqB_trr = 'eqoutB.trr' output_eqB_gro = 'eqoutB.gro' output_eqB_edr = 'eqoutB.edr' output_eqB_log = 'eqoutB.log' # Create and launch bb mdrun(input_tpr_path=output_tprB_eq, output_trr_path=output_eqB_trr, output_gro_path=output_eqB_gro, output_edr_path=output_eqB_edr, output_log_path=output_eqB_log) |
402 403 404 405 406 407 408 409 410 411 412 413 414 415 416 417 418 419 420 421 422 423 424 425 426 427 428 429 | # GMXEnergy: Getting system pressure and density by time during NPT Equilibration from biobb_analysis.gromacs.gmx_energy import gmx_energy #### State A (WT->Mut) #### # Create prop dict and inputs/outputs output_eqA_pd_xvg = 'eqA_PD.xvg' prop = { 'terms': ["Pressure","Density"] } # Create and launch bb gmx_energy(input_energy_path=output_eqA_edr, output_xvg_path=output_eqA_pd_xvg, properties=prop) #### State B (Mut->WT) #### # Create prop dict and inputs/outputs output_eqB_pd_xvg = 'eqB_PD.xvg' prop = { 'terms': ["Pressure","Density"] } # Create and launch bb gmx_energy(input_energy_path=output_eqB_edr, output_xvg_path=output_eqB_pd_xvg, properties=prop) |
433 434 435 436 437 438 439 440 441 442 443 444 445 446 447 448 449 450 451 452 453 454 455 456 457 458 459 460 461 462 463 464 465 466 467 468 469 470 | import plotly from plotly import subplots import plotly.graph_objs as go # Read pressure and density data from file with open(output_eqA_pd_xvg,'r') as pd_file: x,y,z = map( list, zip(*[ (float(line.split()[0]),float(line.split()[1]),float(line.split()[2])) for line in pd_file if not line.startswith(("#","@")) ]) ) plotly.offline.init_notebook_mode(connected=True) trace1 = go.Scatter( x=x,y=y ) trace2 = go.Scatter( x=x,y=z ) fig = subplots.make_subplots(rows=1, cols=2, print_grid=False) fig.append_trace(trace1, 1, 1) fig.append_trace(trace2, 1, 2) fig['layout']['xaxis1'].update(title='Time (ps)') fig['layout']['xaxis2'].update(title='Time (ps)') fig['layout']['yaxis1'].update(title='Pressure (bar)') fig['layout']['yaxis2'].update(title='Density (Kg*m^-3)') fig['layout'].update(title='Pressure and Density during NPT Equilibration') fig['layout'].update(showlegend=False) plotly.offline.iplot(fig) |
474 475 476 477 478 479 480 481 482 483 484 485 486 487 488 489 490 491 492 493 494 495 496 497 498 499 500 501 502 503 504 505 506 507 508 509 510 511 | import plotly from plotly import subplots import plotly.graph_objs as go # Read pressure and density data from file with open(output_eqB_pd_xvg,'r') as pd_file: x,y,z = map( list, zip(*[ (float(line.split()[0]),float(line.split()[1]),float(line.split()[2])) for line in pd_file if not line.startswith(("#","@")) ]) ) plotly.offline.init_notebook_mode(connected=True) trace1 = go.Scatter( x=x,y=y ) trace2 = go.Scatter( x=x,y=z ) fig = subplots.make_subplots(rows=1, cols=2, print_grid=False) fig.append_trace(trace1, 1, 1) fig.append_trace(trace2, 1, 2) fig['layout']['xaxis1'].update(title='Time (ps)') fig['layout']['xaxis2'].update(title='Time (ps)') fig['layout']['yaxis1'].update(title='Pressure (bar)') fig['layout']['yaxis2'].update(title='Density (Kg*m^-3)') fig['layout'].update(title='Pressure and Density during NPT Equilibration') fig['layout'].update(showlegend=False) plotly.offline.iplot(fig) |
515 516 517 518 519 520 521 522 523 524 525 526 527 528 529 530 531 532 533 534 535 536 537 538 539 540 541 542 543 544 545 546 547 548 549 550 551 552 553 554 555 556 557 558 559 560 561 562 563 564 565 566 | # Grompp: Creating portable binary run file for thermodynamic integration (TI) from biobb_gromacs.gromacs.grompp import grompp #### State A (WT->Mut) #### # Create prop dict and inputs/outputs output_tprA_ti = 'tiA.tpr' prop = { 'gmx_lib' : gmxlib, 'mdp':{ 'nsteps':'5000', 'free_energy' : 'yes', 'init-lambda' : '0', 'delta-lambda' : '2e-4', 'sc-alpha' : '0.3', 'sc-coul' : 'yes', 'sc-sigma' : '0.25' }, 'simulation_type': 'free' } # Create and launch bb grompp(input_gro_path=output_eqA_gro, input_top_zip_path=output_pmxtopA_top_zip, output_tpr_path=output_tprA_ti, properties=prop) #### State B (Mut->WT) #### # Create prop dict and inputs/outputs output_tprB_ti = 'tiB.tpr' prop = { 'gmx_lib' : gmxlib, 'mdp':{ 'nsteps':'5000', 'free_energy' : 'yes', 'init-lambda' : '0', 'delta-lambda' : '2e-4', 'sc-alpha' : '0.3', 'sc-coul' : 'yes', 'sc-sigma' : '0.25' }, 'simulation_type': 'free' } # Create and launch bb grompp(input_gro_path=output_eqB_gro, input_top_zip_path=output_pmxtopB_top_zip, output_tpr_path=output_tprB_ti, properties=prop) |
570 571 572 573 574 575 576 577 578 579 580 581 582 583 584 585 586 587 588 589 590 591 592 593 594 595 596 597 598 599 600 601 602 603 604 605 | # Mdrun: Running equilibration from biobb_gromacs.gromacs.mdrun import mdrun #### State A (WT->Mut) #### # Create prop dict and inputs/outputs output_tiA_trr = 'tiA.trr' output_tiA_gro = 'tiA.gro' output_tiA_edr = 'tiA.edr' output_tiA_log = 'tiA.log' output_tiA_dhdl = 'tiA.xvg' # Create and launch bb mdrun(input_tpr_path=output_tprA_ti, output_trr_path=output_tiA_trr, output_gro_path=output_tiA_gro, output_edr_path=output_tiA_edr, output_log_path=output_tiA_log, output_dhdl_path=output_tiA_dhdl) #### State B (Mut->WT) #### # Create prop dict and inputs/outputs output_tiB_trr = 'tiB.trr' output_tiB_gro = 'tiB.gro' output_tiB_edr = 'tiB.edr' output_tiB_log = 'tiB.log' output_tiB_dhdl = 'tiB.xvg' # Create and launch bb mdrun(input_tpr_path=output_tprB_ti, output_trr_path=output_tiB_trr, output_gro_path=output_tiB_gro, output_edr_path=output_tiB_edr, output_log_path=output_tiB_log, output_dhdl_path=output_tiB_dhdl) |
609 610 611 612 613 614 615 616 617 618 619 620 621 622 623 624 625 626 627 628 629 | # Gathering together all the generated dhdl files (work values required to perform the transitions) # from the free energy simulations. # To be used as input for the final pmx free energy estimation. #### State A (WT->Mut) #### zf = zipfile.ZipFile('dhdlsA.zip', mode='w') for file in os.listdir(os.getcwd()): if file.endswith("A.dhdl.xvg"): zf.write(file) zf.close() #### State B (Mut->WT) #### zf = zipfile.ZipFile('dhdlsB.zip', mode='w') for file in os.listdir(os.getcwd()): if file.endswith("B.dhdl.xvg"): zf.write(file) zf.close() |
633 634 635 636 637 638 639 640 641 642 643 644 645 646 647 648 649 650 651 652 653 654 655 656 657 658 659 660 661 | # pmx analyse: analyze_dhdl command from pmx package # Import module from biobb_pmx.pmxbiobb.pmxanalyse import pmxanalyse # Create prop dict and inputs/outputs # Workflow-generated results should be used if a minimum number of transitions are calculated. #state_A_xvg_zip = 'dhdlsA.zip' #state_B_xvg_zip = 'dhdlsB.zip' # In this particular case, as the tutorial is just computing 1 transition (forward + reverse), # values taken from a real run of the snase example will be used instead. state_A_xvg_zip = 'pmx_tutorial/dhdlA.zip' state_B_xvg_zip = 'pmx_tutorial/dhdlB.zip' output_result = 'pmx.txt' output_work_plot = 'pmx.plots.png' prop = { 'reverseB' : True, } #Create and launch bb pmxanalyse(input_a_xvg_zip_path=state_A_xvg_zip, input_b_xvg_zip_path=state_B_xvg_zip, output_result_path=output_result, output_work_plot_path=output_work_plot, properties=prop) |
665 666 | from IPython.display import Image Image(filename=output_work_plot) |
Support
- Share:
-

-

-

- Future updates
Related Workflows





