


Help improve this workflow!
This workflow has been published but could be further improved with some additional meta data:- Keyword(s) in categories input, output, operation, topic
You can help improve this workflow by suggesting the addition or removal of keywords, suggest changes and report issues, or request to become a maintainer of the Workflow .
Repository containing scripts to perform near-real time tracking of SARS-CoV-2 in Connecticut using genomic data. This pipeline is used by the Grubaugh Lab at the Yale School of Public Health, and results are shared on COVIDTrackerCT .
Getting Started
This repository contains scripts for running pre-analyses to prepare sequence and metadata files for running
augur
and
auspice
, and for running the
nextstrain
pipeline itself.
Dependencies
To be able to run the pipeline determined by the
Snakefile
, one needs to set up an extended
conda
nextstrain environment, which will deploy all dependencies (modules and packages) required by the python scripts located at the
scripts
directory. Check each individual script in that directory to know what they do along the workflow.
Setting up a new conda environment
Follow the steps below to set up a conda environment for running the pipeline.
Access a directory or choice in your local machine:
cd 'your/directory/of/choice'
Clone this repository
ncov
git clone https://github.com/andersonbrito/ncov.git
Rename the directory
ncov
as you wish. Access the newly generated directory in your local machine, change directory to
config
, and update your existing nextstrain environment as shown below:
cd 'your/directory/of/choice/ncov/config'
conda env update --file nextstrain.yaml
This command will install all necessary dependencies to run the pipeline, which can be activated by running:
conda activate nextstrain
Preparing the working directory
This minimal set of files and directories are expected in the working directory.
ncov/
│
├── config/
│ ├── auspice_config.json → Auspice configuration file
│ ├── cache_coordinates.tsv → TSV file with pre-existing latitudes and longitudes
│ ├── clades.tsv → TSV file with clade-defining mutations
│ ├── colour_grid.html → HTML file with HEX color matrices
│ ├── dropped_strains.txt → List of genome names to be dropped during the run
│ ├── geoscheme.tsv → Geographic scheme to aggregate locations
│ ├── keep.txt → List of GISAID genomes to be added in the analysis
│ ├── nextstrain.yaml → File used to install nextstrain dependencies
│ ├── reference.gb → GenBank file containing the reference genome annotation
│ └── remove.txt → List of GISAID genomes to be removed prior to the run
│
├── pre-analyses/
│ ├── gisaid_hcov-19.fasta → File with genomes from GISAID (a JSON file can also be used)
│ ├── new_genomes.fasta → FASTA file with the lab's newly sequenced genomes
│ ├── metadata_nextstrain.tsv → nextstrain metadata file, downloaded from GISAID
│ ├── COVID-19_sequencing.xlsx → Custom lab metadata file
│ └── extra_metadata.xlsx → Extra metadata file (can be left blank)
│
├── scripts/ → Set of scripts included in the pipeline
│
├── README.md → Instruction about the pipeline
├── workflow.svg → Diagram showing the pipeline steps
└── Snakefile → Snakemake workflow
Preparing the input data
Files in the
pre-analyses
directory need to be downloaded from distinct sources, as shown below.
| File | Source |
|---|---|
|
gisaid_hcov-19.fasta
provision.json |
FASTA or JSON file downloaded from GISAID |
| new_genomes.fasta¹ | Newly sequenced genomes, with headers formatted as ">Yale-XXX", downloaded from the Lab's Dropbox |
| metadata_nextstrain.tsv² | File 'metadata.tsv' available on GISAID |
| GLab_SC2_sequencing_data.xlsx³ | Metadata spreadsheet downloaded from an internal Google Sheet |
| extra_metadata.xlsx⁴ | Metadata spreadsheet (XLSX) with extra rows, where column names match the main sheet |
Notes:
¹ FASTA file containing all genomes sequenced by the lab, including newly sequenced genomes
² The user will need credentials (login/password) to access and download this file from GISAID
³/⁴ These Excel spreadsheet must have the following columns, named as shown below:
-
Filter → include tags used to filter genomes, as set in
rule filter_metadata -
Sample-ID → lab samples unique identifier, as described below
-
Collection-date
-
Country
-
Division (state) → state name
-
Location (county) → city, town or any other local name
-
Source → lab source of the viral samples
Downloading genome sequences
The files
provision.json
and
gisaid_hcov-19.fasta
contain genomic sequences provided by GISAID. This pipeline takes any of these two file formats as input.
provision.json
can be downloaded via an API provided by GISAID, which require special credentials (username and password, which can be requested via an application process). Contact GISAID Support <[email protected]> for more information.
The file
gisaid_hcov-19.fasta
can be generated via searches on gisaid.org. To do so, the user needs to provide a list of gisaid accession numbers, as follows:
-
The table below illustrates a scheme to sample around 600 genomes of viruses belonging to lineages
B.1.1.7(alpha variant) andB.1.617.2(delta variant), circulating in the US and the United Kingdom, between 2020-12-01 and 2021-06-30, having other US and European samples as contextual genomes. Note that contextual genomes are selected from two time periods, and in different proportions: 50 genomes up to late November 2020, and 100 from December 2020 onwards. Also, the scheme is set up to ignore genomes from California and Scotland, which means that genomes from those locations will not be included in any instance (they are filtered out prior to the genome selection step). To reproduce the scheme above, the following script can be used, having a--metadatafile listing genomes from GISAID that match those filtering categories:
genome_selector.py [-h] --metadata METADATA [--keep KEEP] [--remove REMOVE] --scheme SCHEME [--report REPORT]
... where
--scheme
is a TSV file like the one below:
| purpose | filter | value | filter2 | value2 | sample_size | start | end |
|---|---|---|---|---|---|---|---|
| focus | pango_lineage | B.1.1.7 | country | USA | 200 | 2020-12-01 | 2021-06-30 |
| focus | pango_lineage | B.1.617.2 | country | United Kingdom | 200 | 2020-12-01 | 2021-06-30 |
| context | country | USA | 50 | 2020-11-30 | |||
| context | country | USA | 100 | 2020-12-01 | |||
| context | region | Europe | 100 | 2020-12-01 | 2021-06-30 | ||
| ignore | division | California | |||||
| ignore | division | Scotland |
Among the outputs of
genome_selector.py
users will find text files containing a list of around 550 genome names (e.g. USA/CT-CDC-LC0062417/2021) and a list of gisaid accession numbers (e.g. EPI_ISL_2399048). The first file (with genome names) can be placed in
config/keep.txt
, to list the genomes that will be included in the build. If a file
provision.json
is available, the user can use that as the input sequence file, and that list will be used to retrieve the selected genomes. When the JSON file is not available, the second file (with accession numbers) can be used to filter genomes directly from
gisaid.org
, as follows:
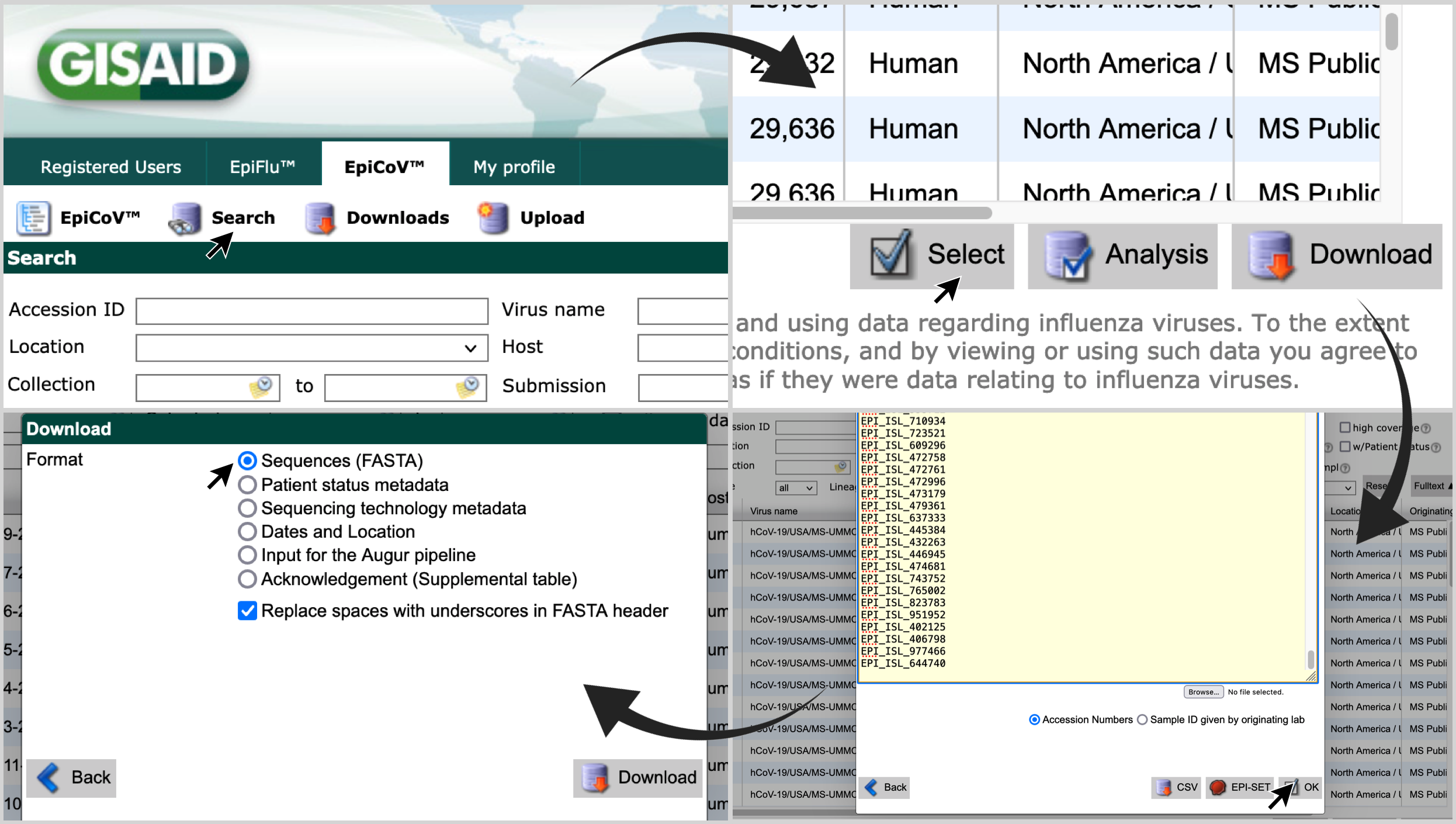
-
Access gisaid.org . Login with your credentials, and click on
Search, andEpiCoV™; -
Click on
Select, paste the list of accession numbers and click onOK; -
Select
Sequences (FASTA)and click onDownload.
The file downloaded via this search can be directly used as the input
pre-analyses/gisaid_hcov-19.fasta
Running the pipeline
Generating augur input data
By running the command below, the appropriate files
sequences.fasta
and
metadata.tsv
will be created inside the
data
directory, and the TSV files
colors.tsv
and
latlongs.tsv
will be created inside the
config
directory:
snakemake preanalyses
Running augur
By running the command below, the rest of the pipeline will be executed:
snakemake export
Removing previous results
By running the command below files related to previous analyses in the working directory will be removed:
snakemake clean
Such command will remove the following files and directories:
results
auspice
data
config/colors.tsv
config/latlongs.tsv
Deleting temporary input files after a successful run
This command will delete the directory
pre-analyses
and its large files:
snakemake delete
New versions
The code in
scripts
will be updated as needed. Re-download this repository (
git clone...
) whenever a new analysis has to be done, to ensure the latest scripts are being used.
Authors
Code Snippets
8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 | import argparse from Bio import SeqIO import json if __name__ == '__main__': parser = argparse.ArgumentParser( description="Append newly sequenced genomes to current genome dataset, and export metadata", formatter_class=argparse.ArgumentDefaultsHelpFormatter ) parser.add_argument("--genomes", required=True, help="FASTA or JSON file with latest genomes from GISAID") parser.add_argument("--new-genomes", required=True, help="FASTA file with newly sequenced genomes") parser.add_argument("--keep", required=True, help="TXT file with accession number of genomes to be included") parser.add_argument("--remove", required=True, help="TXT file with accession number of genomes to be removed") parser.add_argument("--output", required=True, help="FASTA file containing filtered sequences") args = parser.parse_args() genomes = args.genomes new_genomes = args.new_genomes keep = args.keep remove = args.remove outfile = args.output # genomes = path + "pre-analyses/provision.json" # # genomes = path + "pre-analyses/gisaid_hcov-19.fasta" # new_genomes = path + "pre-analyses/new_genomes.fasta" # keep = path + 'config/keep.txt' # remove = path + "config/remove.txt" # outfile = path + "pre-analyses/temp_sequences.fasta" # inspect genome coverage genome_size = 29903 max_gaps = 30 min_size = genome_size - int(genome_size * (max_gaps / 100)) # store only new sequences in a dictionary, ignoring existing ones print('\n### Loading new sequences, and reporting genome coverage\n') newly_sequenced = {} low_coverage = {} for fasta in SeqIO.parse(open(new_genomes), 'fasta'): id, seq = fasta.description, fasta.seq size = len(str(seq).replace('N', '').replace('-', '')) if size > min_size: coverage = str(round(size / genome_size, 3)) print(id + ', coverage = ' + coverage + ' (PASS)') if id not in newly_sequenced.keys(): # avoid potential duplicates newly_sequenced[id] = str(seq) else: coverage = str(round(size / genome_size, 3)) print(id + ', coverage = ' + coverage + ' (FAIL)') low_coverage[id] = coverage print('\nDone!\n') # create a list of sequences to be added in all instances keep_sequences = [] for id in open(keep, "r").readlines(): if id[0] not in ["#", "\n"]: id = id.strip().replace('hCoV-19/', '') if id not in newly_sequenced: if id not in keep_sequences: keep_sequences.append(id) # create a list of sequences to be ignored in all instances remove_sequences = [] for id in open(remove, "r").readlines(): if id[0] not in ["#", "\n"]: id = id.strip() remove_sequences.append(id) # export only sequences to be used in the nextstrain build c = 1 print('\n### Exporting sequences\n') exported = [] ignored = [] all_sequences = [] with open(outfile, 'w') as output: if genomes.split('.')[1] == 'json': with open(genomes) as infile: c = 0 for line in infile: try: entry = json.loads(line) id = entry['covv_virus_name'] id = id.split('|')[0].replace('hCoV-19/', '') if len(id.split('/')) == 3: country, index, year = id.split('/') elif len(id.split('/')) == 4: host, country, index, year = id.split('/') country = country.replace(' ', '').replace('\'', '-').replace('_', '') id = '/'.join([country, index, year]) all_sequences.append(id) if id not in remove_sequences: if id in keep_sequences: # filter out unwanted sequences seq = entry['sequence'].replace('\n', '') if int(len(seq)) >= min_size: entry = ">" + id + "\n" + seq.upper() + "\n" exported.append(id) output.write(entry) print(str(c) + '. ' + id) c += 1 else: ignored.append(id) except: print('Corrupted JSON row detected. Skipping...') else: for fasta in SeqIO.parse(open(genomes), 'fasta'): id, seq = fasta.description, fasta.seq id = id.split('|')[0].replace('hCoV-19/', '') if len(id.split('/')) == 3: country, index, year = id.split('/') elif len(id.split('/')) == 4: host, country, index, year = id.split('/') country = country.replace(' ', '').replace('\'', '-').replace('_', '') id = '/'.join([country, index, year]) all_sequences.append(id) if id not in remove_sequences: if id in keep_sequences: # filter out unwanted sequences entry = ">" + id + "\n" + str(seq).upper() + "\n" exported.append(id) output.write(entry) print(str(c) + '. ' + id) c += 1 else: ignored.append(id) for id, seq in newly_sequenced.items(): print('* ' + str(c) + '. ' + id) entry = ">" + id + "\n" + seq.upper() + "\n" exported.append(id) output.write(entry) c += 1 print('\n- Done!\n') # mismatched sequence headers mismatch = [genome for genome in keep_sequences if genome not in all_sequences] if len(mismatch) + len(low_coverage) > 0: print('\n### WARNINGS!') if len(mismatch) > 0: print('\n### Possible sequence header mismatches\n') m = 1 for id in mismatch: print(str(m) + '. ' + id) m += 1 else: print('\nNo sequence name mismatches found...') if len(low_coverage) > 0: print('\n- Low quality sequences were ignored.\n') l = 1 for id, coverage in low_coverage.items(): print('\t' + str(l) + '. ' + id + ', coverage = ' + coverage + ' (FAIL)') l += 1 print('\n\n\n### Final result\n') print('Lab file contains ' + str(len(newly_sequenced)) + ' high coverage sequences') print('Lab file contains ' + str(len(low_coverage)) + ' low coverage sequences, which were ignored') print('GISAID file contains ' + str(len(all_sequences)) + ' sequences\n') print(str(len(mismatch)) + ' genomes in keep.txt were NOT FOUND on GISAID database') print(str(len(keep_sequences)) + ' genomes ADDED from GISAID dataset') print(str(len(newly_sequenced)) + ' newly sequenced genomes were added') print(str(len(low_coverage)) + ' low coverage genomes were ignored') print(str(len(ignored)) + ' genomes were REMOVED according to remove.txt\n') print(str(len(exported)) + ' genomes included in FINAL dataset\n') |
2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 196 197 198 199 200 201 202 203 204 205 206 207 208 209 210 211 212 213 214 215 216 217 218 219 220 221 222 223 224 225 226 227 228 229 230 231 232 233 234 235 236 237 238 239 240 241 242 243 244 245 246 247 248 249 250 251 252 253 254 255 256 257 258 259 260 261 262 263 264 265 266 267 268 269 270 271 272 273 274 275 276 277 278 279 280 281 282 283 284 285 286 287 288 289 290 291 292 293 294 295 296 297 298 299 300 301 302 303 304 305 306 307 308 309 310 311 312 313 314 315 316 317 318 319 320 321 322 323 324 325 326 327 328 329 330 331 332 333 334 335 336 337 338 339 340 341 342 343 344 345 346 347 348 349 350 351 352 353 354 355 356 357 358 359 360 361 362 363 364 365 366 367 368 369 370 371 372 373 374 375 376 377 378 379 380 381 382 383 384 385 386 387 388 389 390 391 392 393 394 395 396 397 398 399 400 401 402 403 404 405 406 407 408 409 410 411 412 413 414 415 416 417 418 419 420 421 422 423 424 425 426 427 428 429 430 431 432 433 434 435 436 437 438 439 440 441 442 443 444 445 446 447 448 449 450 451 452 453 454 455 456 457 458 459 460 461 462 463 464 465 466 467 468 469 470 471 472 473 474 475 476 477 478 479 480 481 482 483 484 485 486 487 | import pycountry_convert as pyCountry import pandas as pd import argparse from bs4 import BeautifulSoup as BS import pycountry from matplotlib import cm import numpy as np if __name__ == '__main__': parser = argparse.ArgumentParser( description="Generate ordered colour file for nextstrain build", formatter_class=argparse.ArgumentDefaultsHelpFormatter ) parser.add_argument("--metadata", required=True, help="Reformatted nextstrain metadata file") parser.add_argument("--coordinates", required=True, help="TSV coordinates file being used in the build") parser.add_argument("--geoscheme", required=True, help="XML file with geographic scheme") parser.add_argument("--grid", required=True, help="HTML file with HEX colour matrices") parser.add_argument("--columns", nargs='+', type=str, help="list of columns with geographic information") parser.add_argument("--output", required=True, help="TSV file containing ordered HEX colours based on locations") parser.add_argument("--filter", required=False, nargs='+', type=str, help="List of filters for tagged rows in lab metadata") args = parser.parse_args() metadata = args.metadata geoscheme = args.geoscheme coordinates = args.coordinates grid = args.grid columns = args.columns output = args.output filt = args.filter # path = '/Users/anderson/GLab Dropbox/Anderson Brito/projects/ncov/ncov_variants/nextstrain/runX_ncov_20210407_vocvoicolours/' # metadata = path + 'data/metadata.tsv' # coordinates = path + 'config/latlongs.tsv' # geoscheme = path + 'config/geoscheme.tsv' # grid = path + 'config/colour_grid.html' # columns = ['region', 'country', 'division', 'location'] # output = path + 'data/colors.tsv' # pre-determined HEX colours and hues print(filt) if filt==['connecticut']: force_colour = {'Connecticut': '#8FEECB', 'New York': '#19ae77', 'Canada': '#663300'} force_hue = {'North America': 0} if filt==['caribbean']: force_colour = {'Puerto Rico': '#8FEECB', 'US Virgin Islands': '#19ae77', 'Dominican Republic': '#663300'} force_hue = {'Caribbean': 0} # content to be exported as final result latlongs = {trait: {} for trait in columns} # get ISO alpha3 country codes isos = {'US Virgin Islands':'VIR','Virgin Islands':'VIR','British Virgin Islands':'VGB','Curacao':'CUW','Northern Mariana Islands':'MNP', 'Sint Maarten':'MAF','St Eustatius':'BES'} def get_iso(country): global isos if country not in isos.keys(): try: isoCode = pyCountry.country_name_to_country_alpha3(country, cn_name_format="default") isos[country] = isoCode except: try: isoCode = pycountry.countries.search_fuzzy(country)[0].alpha_3 isos[country] = isoCode except: isos[country] = '' return isos[country] # extract coordinates from latlongs file for sorting places by latitude for line in open(coordinates).readlines(): if not line.startswith('\n'): try: trait, place, lat, long = line.strip().split('\t') if trait in latlongs.keys(): entry = {place: (str(lat), str(long))} latlongs[trait].update(entry) # save as pre-existing result else: print('### WARNING! ' + trait + ' is not among the pre-selected columns with geographic information!') except: pass ''' REORDER LOCATIONS FOR LEGEND FORMATTING ''' # open metadata file as dataframe dfN = pd.read_csv(metadata, encoding='utf-8', sep='\t', dtype=str) df = dfN dfN = dfN[['region', 'country', 'division', 'location']] ordered_regions = {} dcountries = {} places = [] pinpoints = [dfN[trait].values.tolist() for trait in columns] for region_index in latlongs[columns[0]]: for address in zip(*pinpoints): address = list(address) region, country, division, location = [trait for trait in columns] region_name = address[0] country_name = address[1] division_name = address[2] location_name = address[3] places.append(address) if region_index == region_name: if 'subcontinent' not in ordered_regions.keys(): ordered_regions['subcontinent'] = {} if region_name not in ordered_regions['subcontinent'].keys(): ordered_regions['subcontinent'].update({region_name: latlongs[region][region_name]}) ### STORE COUNTRIES WITH REGIONS AS KEYS if country_name in latlongs[country].keys(): if region_name not in dcountries.keys(): dcountries[region_name] = {country_name: latlongs[country][country_name]} else: if country_name not in dcountries[region_name].keys(): dcountries[region_name].update({country_name: latlongs[country][country_name]}) # sort division entries based on sorted country entries ordered_countries = {} for region, countries in dcountries.items(): ordered_countries[region] = {k: v for k, v in sorted(countries.items(), key=lambda item: item[1])} ddivisions = {} for country_index in [key for dict_ in ordered_countries.values() for key in dict_]: for address in places: region, country, division, location = [trait for trait in columns] country_name = address[1] division_name = address[2] location_name = address[3] if country_index == country_name: ### STORE DIVISIONS WITH COUNTRIES AS KEYS if division_name in latlongs[division].keys(): if country_name not in ddivisions.keys(): ddivisions[country_name] = {division_name: latlongs[division][division_name]} else: if division_name not in ddivisions[country_name].keys(): ddivisions[country_name].update({division_name: latlongs[division][division_name]}) # sort division entries based on sorted country entries ordered_divisions = {} for country, divisions in ddivisions.items(): ordered_divisions[country] = {k: v for k, v in sorted(divisions.items(), key=lambda item: item[1])} dlocations = {} for division_index in [key for dict_ in ordered_divisions.values() for key in dict_]: for address in places: address = list(address) region, country, division, location = [trait for trait in columns] division_name = address[2] location_name = address[3] if division_index == division_name: ### STORE LOCATIONS WITH DIVISIONS AS KEYS if location_name in latlongs[location].keys(): if division_name not in dlocations.keys(): dlocations[division_name] = {location_name: latlongs[location][location_name]} else: if location_name not in dlocations[division_name].keys(): dlocations[division_name].update({location_name: latlongs[location][location_name]}) # sort locations entries based on sorted division entries ordered_locations = {} for division, locations in dlocations.items(): ordered_locations[division] = {k: v for k, v in sorted(locations.items(), key=lambda item: item[1])} ''' CONVERT RGB TO HEX COLOURS ''' # original source: https://bsou.io/posts/color-gradients-with-python # convert colour codes def RGB_to_hex(RGB): ''' [255,255,255] -> "#FFFFFF" ''' # Components need to be integers for hex to make sense RGB = [int(x) for x in RGB] return "#" + "".join(["0{0:x}".format(v) if v < 16 else "{0:x}".format(v) for v in RGB]) def hex_to_RGB(hex): ''' "#FFFFFF" -> [255,255,255] ''' # Pass 16 to the integer function for change of base return [int(hex[i:i + 2], 16) for i in range(1, 6, 2)] def color_dict(gradient): ''' Takes in a list of RGB sub-lists and returns dictionary of colors in RGB and hex form for use in a graphing function defined later on ''' return [RGB_to_hex(RGB) for RGB in gradient] # create gradient def linear_gradient(start_hex, finish_hex, n): ''' returns a gradient list of (n) colors between two hex colors. start_hex and finish_hex should be the full six-digit color string, ("#FFFFFF") ''' # Starting and ending colors in RGB form s = hex_to_RGB(start_hex) f = hex_to_RGB(finish_hex) # Initilize a list of the output colors with the starting color RGB_list = [s] # Calcuate a color at each evenly spaced value of t from 1 to n n = n for t in range(1, n): # Interpolate RGB vector for color at the current value of t curr_vector = [ int(s[j] + (float(t) / (n - 1)) * (f[j] - s[j])) for j in range(3)] # Add it to list of output colors RGB_list.append(curr_vector) RGB_list = RGB_list return color_dict(RGB_list) ''' IMPORT GEOSCHEME ''' # xml = BS(open(geoscheme, "r").read(), 'xml') # levels = xml.find('levels') scheme_list = open(geoscheme, "r").readlines()[1:] sampled_region = [key for dict_ in ordered_regions.values() for key in dict_] geodata = {} for line in scheme_list: if not line.startswith('\n'): type = line.split('\t')[0] if type == 'region': continent = line.split('\t')[1] region = line.split('\t')[2] if region in sampled_region: if continent not in geodata.keys(): geodata[continent] = [region] else: geodata[continent] += [region] ''' IMPORT COLOUR SCHEME ''' print('\nGenerating colour scheme...\n') html = BS(open(grid, "r").read(), 'html.parser') tables = html.find_all('table') # for colour_name in colour_scale.keys(): limits = {'dark': (60, 30), 'light': (100, 80)} # define saturation and luminance, max and min, respectively hue_to_hex = {} for table in tables: string_head = str(table.caption) if string_head == 'None': continue else: hue_value = int(string_head.split('"')[1]) lux = table.tbody.find_all('tr') lum_value = 90 # brightest colour hexdark = '' hexligth = '' for row in lux: sat_value = 10 # unsaturated colour for cell in row.find_all('td'): # pass if sat_value == limits['dark'][0] and lum_value == limits['dark'][1]: hexdark = cell.text.strip() if sat_value == limits['light'][0] and lum_value == limits['light'][1]: hexligth = cell.text.strip() hex = (hexdark, hexligth) sat_value += 10 lum_value -= 10 hex = (hexdark, hexligth) hue_to_hex[hue_value] = hex colour_scale = {'magenta': [320], 'purple': [310, 300, 290, 280, 270, 260], 'blue': [250, 240, 230, 220], 'cyan': [210, 200, 190, 180], 'turquoise': [170, 160, 150], 'green': [140, 130, 120], 'yellowgreen': [110, 100, 90, 80, 70], 'yellow': [60, 50, 40], 'orange': [30, 20], 'red': [10, 0]} continent_hues = {'Oceania': colour_scale['magenta'], 'Asia': colour_scale['purple'], 'Europe': colour_scale['blue'] + colour_scale['cyan'], 'Africa': colour_scale['yellowgreen'], 'America': colour_scale['yellow'] + colour_scale['orange'] + colour_scale['red']} colour_wheel = {} palette = {} for area, subareas in geodata.items(): num_subareas = len(subareas) hues = len(continent_hues[area]) print(area, subareas) for position, subarea in zip([int(x) for x in np.linspace(0, int(hues), num_subareas, endpoint=False)], subareas): if subarea not in palette.keys(): hue = continent_hues[area][position] # colour picker palette[subarea] = hue # print(subarea, hue) print(colour_wheel) # print(sampled_region) # print(palette) for region in sampled_region: if region in force_hue: colour_wheel[region] = hue_to_hex[force_hue[region]] else: colour_wheel[region] = hue_to_hex[palette[region]] ''' SET COLOUR SCHEME FOR UPDATES ''' # convert a hue value into an rgb colour, and then hex colour def hue_to_rgb(hue): colour = int(hue / 240 * 255) # print(colour) luminance = 0.7 rgb = [c * 255 * luminance for c in list(cm.jet(colour))[:-1]] # print(rgb) return RGB_to_hex(rgb) results = {trait: {} for trait in columns} ''' APPLY SAME HUE FOR MEMBERS OF THE SAME SUB-CONTINENT ''' # assign countries to regions country_colours = {} reference_countries = {} for region, members in ordered_countries.items(): countries = list(members.keys()) for country in countries: reference_countries[country] = region country_colours[region] = countries # assign divisions to countries division_colours = {} reference_divisions = {} for country, members in ordered_divisions.items(): divisions = list(members.keys()) for division in divisions: reference_divisions[division] = reference_countries[country] if reference_countries[country] not in division_colours.keys(): division_colours[reference_countries[country]] = divisions else: if division not in division_colours[reference_countries[country]]: division_colours[reference_countries[country]].append(division) # assign locations to divisions location_colours = {} reference_locations = {} for division, members in ordered_locations.items(): locations = list(members.keys()) for location in locations: if reference_divisions[division] not in location_colours.keys(): location_colours[reference_divisions[division]] = locations else: if location not in location_colours[reference_divisions[division]]: location_colours[reference_divisions[division]].append(location) ''' CREATE COLOUR GRADIENT ''' # define gradients for regions for continent, regions in geodata.items(): hex_limits = [] for region in sampled_region: if region in regions: if continent == 'America': hex_limits += [list(colour_wheel[region])[0]] else: hex_limits += list(colour_wheel[region]) start, end = hex_limits[0], hex_limits[-1] if len(regions) == 1: gradient = linear_gradient(start, end, 4) gradient = [list(gradient)[2]] else: gradient = linear_gradient(start, end, len(regions)) for region, colour in zip(regions, gradient): print('region', region, colour) results['region'].update({region: colour}) # define gradients for country for hue, countries in country_colours.items(): start, end = colour_wheel[hue] if len(countries) == 1: gradient = linear_gradient(start, end, 4) gradient = [list(gradient)[2]] else: gradient = linear_gradient(start, end, len(countries)) for country, colour in zip(countries, gradient): print('country', country, colour) results['country'].update({country: colour}) # define gradients for divisions for hue, divisions in division_colours.items(): start, end = colour_wheel[hue] if len(divisions) == 1: gradient = linear_gradient(start, end, 3) gradient = [list(gradient)[1]] else: gradient = linear_gradient(start, end, len(divisions)) for division, colour in zip(divisions, gradient): print('division', division, colour) results['division'].update({division: colour}) # define gradients for locations for hue, locations in location_colours.items(): start, end = colour_wheel[hue] if len(locations) == 1: gradient = linear_gradient(start, end, 3) gradient = [list(gradient)[1]] else: gradient = linear_gradient(start, end, len(locations)) for location, colour in zip(locations, gradient): print('location', location, colour) results['location'].update({location: colour}) # special colouring geoLevels = {} for line in scheme_list: if not line.startswith('\n'): line = line.strip() id = line.split('\t')[2] type = line.split('\t')[0] # parse subnational regions for countries in geoscheme if type == 'country': members = [item.strip() for item in line.split('\t')[5].split(',')] # elements inside the subarea if id not in geoLevels: geoLevels[id] = members categories = {'Global': '#CCCCCC', 'Europe': '#666666'} results['us_region'] = {} usregion_hues = { 'USA-Northeast': colour_scale['purple'][3], 'USA-Midwest': colour_scale['green'][0], 'USA-Southwest': colour_scale['yellow'][1], 'USA-Southeast': colour_scale['cyan'][0], 'USA-West': colour_scale['red'][0] } for us_region, hue in usregion_hues.items(): start, end = hue_to_hex[hue] divisions = geoLevels[us_region] gradient = linear_gradient(start, end, len(divisions)) # print(us_region, hue, divisions, gradient) for state, colour in zip(divisions, gradient): categories[state] = colour for reg, hex in categories.items(): results['us_region'].update({reg: hex}) print('us_region', reg, hex) # VOC / VOI list_category = [up_number for up_number in sorted(set(df['category'].to_list())) if up_number != 'Other variants'] list_hex = list([hue_to_rgb(int(x)) for x in np.linspace(30, 240, len(list_category)*2, endpoint=True)]) skip_hex = [h for n, h in enumerate(list_hex) if n in range(0, len(list_hex), 2)][::-1] results['category'] = {} for category, hex in zip(list_category, skip_hex): results['category'].update({category: hex}) print(category, hex) results['category'].update({'Other variants': '#808080'}) ''' EXPORT COLOUR FILE ''' with open(output, 'w') as outfile: for trait, entries in results.items(): for place, hexcolour in entries.items(): if place in force_colour and trait not in ['location']: hexcolour = force_colour[place] print('* ' + place + ' is hardcode with the colour ' + hexcolour) line = "{}\t{}\t{}\n".format(trait, place, hexcolour.upper()) outfile.write(line) outfile.write('\n') print('\nOrdered colour file successfully created!\n') |
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 | import pycountry_convert as pyCountry import pycountry import pandas as pd import argparse from uszipcode import SearchEngine if __name__ == '__main__': parser = argparse.ArgumentParser( description="Reformat metadata file by adding column with subcontinental regions based on the UN geo-scheme", formatter_class=argparse.ArgumentDefaultsHelpFormatter ) parser.add_argument("--metadata", required=True, help="Nextstrain metadata file") parser.add_argument("--geoscheme", required=True, help="XML file with geographic classifications") parser.add_argument("--output", required=True, help="Updated metadata file") parser.add_argument("--filter", required=True, nargs='+', type=str, help="Filter region to define focus") args = parser.parse_args() metadata = args.metadata geoscheme = args.geoscheme output = args.output filt = args.filter # path = '/Users/anderson/GLab Dropbox/Anderson Brito/projects/ncov/ncov_variants/nextstrain/run6_20210202_b117/ncov/' # metadata = path + 'pre-analyses/metadata_filtered.tsv' # geoscheme = path + 'config/geoscheme.tsv' # output = path + 'pre-analyses/metadata_geo.tsv' # ##keep location (county) data for focus region of build if filt==['caribbean']: focus = ['US Virgin Islands','Puerto Rico','Dominican Republic','Anguilla','Antigua and Barbuda', 'Aruba','Bahamas','Barbados','Bonaire','Sint Eustatius','Saba','British Virgin Islands','Cayman Islands', 'Cuba','Curacao','Dominica','Grenada','Guadeloupe','Haiti','Jamaica','Martinique','Montserrat', 'Saint Barthelemy','Saint Kitts and Nevis','Saint Lucia','Saint Martin','Saint Vincent and the Grenadines', 'Sint Maarten','Trinidad and Tobago','Turks and Caicos', 'USA','Netherlands','United Kingdom','France'] if filt == ['connecticut']: focus = ['USA', 'Canada', 'United Kingdom', 'Maine', 'New Hampshire', 'Massachusetts', 'Connecticut', 'Vermont', 'New York'] if filt == ['']: focus = [] print(focus) # get ISO alpha3 country codes isos = {'British Virgin Islands':'VGB','Cayman Islands':'CYM','Guam':'GUM','US Virgin Islands':'VIR','Virgin Islands':'VIR', 'Puerto Rico':'PRI','Northern Mariana Islands':'MNP','Sint Maarten':'MAF','Sint Eustatius':'BES','St Eustatius':'BES', 'Bonaire':'BES','Saba':'BES','Martinique':'MTQ','French Guiana':'GUF','Montserrat':'MSR','Turks and Caicos':'TCA', 'Anguilla':'AIA','Mayotte':'MYT','Reunion':'RUE','Wallis and Futuna':'WLF'} def get_iso(country): global isos if country not in isos.keys(): try: isoCode = pyCountry.country_name_to_country_alpha3(country, cn_name_format="default") isos[country] = isoCode except: try: isoCode = pycountry.countries.search_fuzzy(country)[0].alpha_3 isos[country] = isoCode except: isos[country] = '' return isos[country] #keys to correct ISOs for colonies colonies = {'British Virgin Islands':'VGB','Cayman Islands':'CYM','Guam':'GUM','US Virgin Islands':'VIR','Virgin Islands':'VIR', 'Puerto Rico':'PRI','Northern Mariana Islands':'MNP','Sint Maarten':'MAF','Sint Eustatius':'BES','St Eustatius':'BES', 'Bonaire':'BES','Saba':'BES','Martinique':'MTQ','French Guiana':'GUF','Montserrat':'MSR','Turks and Caicos':'TCA', 'Anguilla':'AIA','Mayotte':'MYT','Reunion':'RUE','Wallis and Futuna':'WLF'} #keys to correct regions for colonies colony_regions = {'British Virgin Islands':'Caribbean','Cayman Islands':'Caribbean','Guam':'Oceania','US Virgin Islands':'Caribbean','Virgin Islands':'Caribbean', 'Puerto Rico':'Caribbean','Northern Mariana Islands':'Oceania','Sint Maarten':'Caribbean','Sint Eustatius':'Caribbean','St Eustatius':'Caribbean', 'Bonaire':'Caribbean','Saba':'Caribbean','Martinique':'Caribbean','French Guiana':'South America','Montserrat':'Caribbean','Turks and Caicos':'Caribbean', 'Anguilla':'Caribbean','Mayotte':'Eastern Africa','Reunion':'Eastern Africa','Wallis and Futuna':'Oceania'} #keys to correct common misspellings (in GISAID or in our metadata) misspelled = {'Virgin Islands':'US Virgin Islands','St Eustatius':'Sint Eustatius','Turks and Caicos Islands':'Turks and Caicos'} # parse subcontinental regions in geoscheme scheme_list = open(geoscheme, "r").readlines()[1:] geoLevels = {} c = 0 for line in scheme_list: if not line.startswith('\n'): id = line.split('\t')[2] type = line.split('\t')[0] if type == 'region': members = line.split('\t')[5].split(',') # elements inside the subarea for country in members: iso = get_iso(country.strip()) geoLevels[iso] = id # parse subnational regions for countries in geoscheme if type == 'country': members = line.split('\t')[5].split(',') # elements inside the subarea for state in members: if state.strip() not in geoLevels.keys(): geoLevels[state.strip()] = id # parse subareas for states in geoscheme if type == 'location': members = line.split('\t')[5].split(',') # elements inside the subarea for zipcode in members: if zipcode.strip() not in geoLevels.keys(): geoLevels[zipcode.strip()] = id # open metadata file as dataframe dfN = pd.read_csv(metadata, encoding='utf-8', sep='\t') try: dfN.insert(4, 'region', '') except: pass dfN['region'] = dfN['iso'].map(geoLevels) # add 'column' region in metadata dfN['us_region'] = '' listA = dfN['iso'] + dfN['iso'].map(geoLevels) listA.to_csv('listA.csv', sep='\t', index=False) # convert sets of locations into sub-locations print('\nApplying geo-schemes...') dfN.fillna('', inplace=True) for idx, row in dfN.iterrows(): # flatten divison names as country names, for countries that are not a focus of study country = dfN.loc[idx, 'country'] division = dfN.loc[idx,'division'] if country not in focus: if division not in focus: dfN.loc[idx, 'division'] = country # assign US region if country not in ['USA']: if 'Europe' in dfN.loc[idx, 'region']: dfN.loc[idx, 'us_region'] = 'Europe' else: dfN.loc[idx, 'us_region'] = 'Global' if country == 'USA' and dfN.loc[idx, 'us_region'] == '': dfN.loc[idx, 'us_region'] = dfN.loc[idx, 'division'] # divide country into subnational regions division = dfN.loc[idx, 'division'] if division not in ['', 'unknown']: if division in geoLevels.keys(): dfN.loc[idx, 'country'] = geoLevels[dfN.loc[idx, 'division']] # correct ISO codes and regions for colonies (any insular terrritories) island = dfN.loc[idx,'country'] if island in colonies.keys(): dfN.loc[idx,'iso'] = colonies[island] dfN.loc[idx,'region'] = colony_regions[island] # flatten location names as division names for divisions that are not a focus of study if division not in focus: dfN.loc[idx, 'location'] = division #print('Processing metadata for... ' + row['strain']) #rename some commonly misspelled or mismatched data if country in misspelled: dfN.loc[idx,'country'] = misspelled[country] if division in misspelled: dfN.loc[idx,'division'] = misspelled[division] location = dfN.loc[idx,'location'] if location in misspelled: dfN.loc[idx,'location'] = misspelled[location] #rename Florida, PR if dfN.loc[idx,'country'] in ['Puerto Rico']: if dfN.loc[idx,'location'] in ['Florida']: dfN.loc[idx,'location'] = 'Florida (PR)' dfN = dfN.drop_duplicates(subset=['strain']) dfN.to_csv(output, sep='\t', index=False) print('\nMetadata file successfully reformatted applying geo-scheme!\n') |
9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 196 197 198 199 200 201 202 203 204 205 206 207 208 209 210 211 212 213 214 215 216 217 218 219 220 221 222 223 224 225 226 227 228 229 230 231 232 233 234 235 236 237 238 239 240 241 242 243 244 245 246 247 248 249 250 251 252 253 254 255 256 257 258 259 260 261 262 263 264 265 266 267 268 269 270 271 272 273 274 275 276 277 278 279 280 281 282 283 284 285 286 287 288 289 290 291 292 293 294 295 296 297 298 299 300 301 302 303 304 305 306 307 308 309 310 311 312 313 314 315 316 317 318 319 320 321 322 323 324 325 326 327 328 329 330 331 332 333 334 335 336 337 338 339 340 341 342 343 344 345 346 347 348 349 350 351 352 353 354 355 356 357 358 359 360 361 362 363 364 365 366 367 368 369 370 371 372 373 374 375 376 377 378 379 380 381 382 383 384 385 386 387 388 389 390 391 392 393 394 395 396 397 398 399 400 401 402 403 404 405 406 407 408 409 410 411 412 413 414 415 416 417 418 419 420 421 422 423 424 425 426 427 428 429 430 431 432 433 434 435 436 437 438 439 440 441 442 443 444 445 446 447 448 449 450 451 452 453 454 455 456 457 458 459 460 461 462 463 464 465 466 467 468 469 470 471 472 473 474 475 476 477 478 479 480 481 482 483 484 485 486 487 488 489 490 491 492 493 494 495 496 497 498 499 500 501 502 503 504 505 506 507 508 509 510 511 512 513 514 515 516 517 518 519 520 521 522 523 524 525 526 527 528 529 530 531 532 533 534 535 536 | import pycountry_convert as pyCountry import pycountry from Bio import SeqIO import pandas as pd from epiweeks import Week import time import argparse if __name__ == '__main__': parser = argparse.ArgumentParser( description="Filter nextstrain metadata files re-formmating and exporting only selected lines", formatter_class=argparse.ArgumentDefaultsHelpFormatter ) parser.add_argument("--genomes", required=True, help="FASTA file genomes to be used") parser.add_argument("--metadata1", required=True, help="Metadata file from NextStrain") parser.add_argument("--metadata2", required=False, help="Custom lab metadata file") parser.add_argument("--filter", required=False, nargs='+', type=str, help="List of filters for tagged rows in lab metadata") parser.add_argument("--output1", required=True, help="Filtered metadata file") parser.add_argument("--output2", required=True, help="Reformatted, final FASTA file") args = parser.parse_args() genomes = args.genomes metadata1 = args.metadata1 metadata2 = args.metadata2 filterby = args.filter output1 = args.output1 output2 = args.output2 # path = '/Users/anderson/GLab Dropbox/Anderson Brito/projects/ncov/ncov_variants/nextstrain/runX_20210617_filter/' # genomes = path + 'pre-analyses/temp_sequences.fasta' # metadata1 = path + 'pre-analyses/metadata_nextstrain.tsv' # metadata2 = path + 'pre-analyses/GLab_SC2_sequencing_data.xlsx' # filterby = ['caribe', 'test'] # output1 = path + 'pre-analyses/metadata_filtered.tsv' # output2 = path + 'pre-analyses/sequences.fasta' # temporal boundaries today = time.strftime('%Y-%m-%d', time.gmtime()) min_date = '2019-12-15' variants = {'B.1.1.7': 'Alpha (B.1.1.7)', 'B.1.351': 'Beta (B.1.351)', 'B.1.351.2': 'Beta (B.1.351.2)', 'B.1.351.3': 'Beta (B.1.351.3)', 'P.1': 'Gamma (P.1)', 'P.1.1': 'Gamma (P.1.1)', 'P.1.2': 'Gamma (P.1.2)', 'B.1.617.2': 'Delta (B.1.617.2)', 'AY.1': 'Delta (AY.1)', 'AY.2': 'Delta (AY.2)', 'AY.3': 'Delta (AY.3)', 'AY.3.1': 'Delta (AY.3.1)', 'AY.4': 'Delta (AY.4)', 'AY.4.1': 'Delta (AY.4.1)', 'AY.4.2': 'Delta (AY.4.2)', 'AY.4.3': 'Delta (AY.4.3)', 'AY.4.4': 'Delta (AY.4.4)', 'AY.4.5': 'Delta (AY.4.5)', 'AY.5': 'Delta (AY.5)', 'AY.5.1': 'Delta (AY.5.1)', 'AY.5.2': 'Delta (AY.5.2)', 'AY.5.3': 'Delta (AY.5.3)', 'AY.5.4': 'Delta (AY.5.4)', 'AY.6': 'Delta (AY.6)', 'AY.7': 'Delta (AY.7)', 'AY.7.1': 'Delta (AY.7.1)', 'AY.7.2': 'Delta (AY.7.2)', 'AY.8': 'Delta (AY.8)', 'AY.9': 'Delta (AY.9)', 'AY.9.1': 'Delta (AY.9.1)', 'AY.9.2': 'Delta (AY.9.2)', 'AY.9.2.1': 'Delta (AY.9.2.1)', 'AY.10': 'Delta (AY.10)', 'AY.11': 'Delta (AY.11)', 'AY.12': 'Delta (AY.12)', 'AY.13': 'Delta (AY.13)', 'AY.14': 'Delta (AY.14)', 'AY.15': 'Delta (AY.15)', 'AY.16': 'Delta (AY.16)', 'AY.16.1': 'Delta (AY.16.1)', 'AY.17': 'Delta (AY.17)', 'AY.18': 'Delta (AY.18)', 'AY.19': 'Delta (AY.19)', 'AY.20': 'Delta (AY.20)', 'AY.21': 'Delta (AY.21)', 'AY.22': 'Delta (AY.22)', 'AY.23': 'Delta (AY.23)', 'AY.23.1': 'Delta (AY.23.1)', 'AY.24': 'Delta (AY.24)', 'AY.25': 'Delta (AY.25)', 'AY.26': 'Delta (AY.26)', 'AY.27': 'Delta (AY.27)', 'AY.28': 'Delta (AY.28)', 'AY.29': 'Delta (AY.29)', 'AY.29.1': 'Delta (AY.29.1)', 'AY.30': 'Delta (AY.30)', 'AY.31': 'Delta (AY.31)', 'AY.32': 'Delta (AY.32)', 'AY.33': 'Delta (AY.33)', 'AY.34': 'Delta (AY.34)', 'AY.35': 'Delta (AY.35)', 'AY.36': 'Delta (AY.36)', 'AY.37': 'Delta (AY.37)', 'AY.38': 'Delta (AY.38)', 'AY.39': 'Delta (AY.39)', 'AY.39.1': 'Delta (AY.39.1)', 'AY.39.1.1': 'Delta (AY.39.1.1)', 'AY.39.2': 'Delta (AY.39.2)', 'AY.40': 'Delta (AY.40)', 'AY.41': 'Delta (AY.41)', 'AY.42': 'Delta (AY.42)', 'AY.43': 'Delta (AY.43)', 'AY.44': 'Delta (AY.44)', 'AY.45': 'Delta (AY.45)', 'AY.46': 'Delta (AY.46)', 'AY.46.1': 'Delta (AY.46.1)', 'AY.46.2': 'Delta (AY.46.2)', 'AY.46.3': 'Delta (AY.46.3)', 'AY.46.4': 'Delta (AY.46.4)', 'AY.46.5': 'Delta (AY.46.5)', 'AY.46.6': 'Delta (AY.46.6)', 'AY.47': 'Delta (AY.47)', 'AY.48': 'Delta (AY.48)', 'AY.49': 'Delta (AY.49)', 'AY.50': 'Delta (AY.50)', 'AY.51': 'Delta (AY.51)', 'AY.52': 'Delta (AY.52)', 'AY.53': 'Delta (AY.53)', 'AY.54': 'Delta (AY.54)', 'AY.55': 'Delta (AY.55)', 'AY.56': 'Delta (AY.56)', 'AY.57': 'Delta (AY.57)', 'AY.58': 'Delta (AY.58)', 'AY.59': 'Delta (AY.59)', 'AY.60': 'Delta (AY.60)', 'AY.61': 'Delta (AY.61)', 'AY.62': 'Delta (AY.62)', 'AY.63': 'Delta (AY.63)', 'AY.64': 'Delta (AY.64)', 'AY.65': 'Delta (AY.65)', 'AY.66': 'Delta (AY.66)', 'AY.67': 'Delta (AY.67)', 'AY.68': 'Delta (AY.68)', 'AY.69': 'Delta (AY.69)', 'AY.70': 'Delta (AY.70)', 'AY.71': 'Delta (AY.71)', 'AY.72': 'Delta (AY.72)', 'AY.73': 'Delta (AY.73)', 'AY.74': 'Delta (AY.74)', 'AY.75': 'Delta (AY.75)', 'AY.75.1': 'Delta (AY.75.1)', 'AY.76': 'Delta (AY.76)', 'AY.77': 'Delta (AY.77)', 'AY.78': 'Delta (AY.78)', 'AY.79': 'Delta (AY.79)', 'AY.80': 'Delta (AY.80)', 'AY.81': 'Delta (AY.81)', 'AY.82': 'Delta (AY.82)', 'AY.83': 'Delta (AY.83)', 'AY.84': 'Delta (AY.84)', 'AY.85': 'Delta (AY.85)', 'AY.86': 'Delta (AY.86)', 'AY.87': 'Delta (AY.87)', 'AY.88': 'Delta (AY.88)', 'AY.89': 'Delta (AY.89)', 'AY.90': 'Delta (AY.90)', 'AY.91': 'Delta (AY.91)', 'AY.91.1': 'Delta (AY.91.1)', 'AY.92': 'Delta (AY.92)', 'AY.93': 'Delta (AY.93)', 'AY.94': 'Delta (AY.94)', 'AY.95': 'Delta (AY.95)', 'AY.96': 'Delta (AY.96)', 'AY.97': 'Delta (AY.97)', 'AY.98': 'Delta (AY.98)', 'AY.98.1': 'Delta (AY.98.1)', 'AY.99': 'Delta (AY.99)', 'AY.99.1': 'Delta (AY.99.1)', 'AY.99.2': 'Delta (AY.99.2)', 'AY.100': 'Delta (AY.100)', 'AY.101': 'Delta (AY.101)', 'AY.102': 'Delta (AY.102)', 'AY.103': 'Delta (AY.103)', 'AY.104': 'Delta (AY.104)', 'AY.105': 'Delta (AY.105)', 'AY.106': 'Delta (AY.106)', 'AY.107': 'Delta (AY.107)', 'AY.108': 'Delta (AY.108)', 'AY.109': 'Delta (AY.109)', 'AY.110': 'Delta (AY.110)', 'AY.111': 'Delta (AY.111)', 'AY.112': 'Delta (AY.112)', 'AY.113': 'Delta (AY.113)', 'AY.114': 'Delta (AY.114)', 'AY.115': 'Delta (AY.115)', 'AY.116': 'Delta (AY.116)', 'AY.116.1': 'Delta (AY.116.1)', 'AY.117': 'Delta (AY.117)', 'AY.118': 'Delta (AY.118)', 'AY.119': 'Delta (AY.119)', 'AY.120': 'Delta (AY.120)', 'AY.120.1': 'Delta (AY.120.1)', 'AY.120.2': 'Delta (AY.120.2)', 'AY.120.2.1': 'Delta (AY.120.2.1)', 'AY.121': 'Delta (AY.121)', 'AY.121.1': 'Delta (AY.121.1)', 'AY.122': 'Delta (AY.122)', 'AY.122.1': 'Delta (AY.122.1)', 'AY.123': 'Delta (AY.123)', 'AY.124': 'Delta (AY.124)', 'AY.125': 'Delta (AY.125)', 'AY.126': 'Delta (AY.126)', 'AY.127': 'Delta (AY.127)', 'AY.128': 'Delta (AY.128)', 'AY.129': 'Delta (AY.129', 'AY.130': 'Delta (AY.130)', 'B.1.525': 'Eta (B.1.525)', 'B.1.526': 'Iota (B.1.526)', 'B.1.617.1': 'Kappa (B.1.617.1)', 'C.37': 'Lambda (C.37)', 'B.1.427': 'Epsilon (B.1.427/B.1.429)', 'B.1.429': 'Epsilon (B.1.427/B.1.429)', 'P.2': 'Zeta (P.2)', 'B.1.621': 'Mu (B.1.621)', 'B.1.621.1': 'Mu (B.1.621.1)', 'BA.1': 'Omicron (BA.1)' } # get ISO alpha3 country codes isos = {'US Virgin Islands':'VIR','Virgin Islands':'VIR','British Virgin Islands':'VGB','Curacao':'CUW','Northern Mariana Islands':'MNP', 'Sint Maarten':'MAF','St Eustatius':'BES'} def get_iso(country): global isos if country not in isos.keys(): try: isoCode = pyCountry.country_name_to_country_alpha3(country, cn_name_format="default") isos[country] = isoCode except: try: isoCode = pycountry.countries.search_fuzzy(country)[0].alpha_3 isos[country] = isoCode except: isos[country] = '' return isos[country] # create epiweek column def get_epiweeks(date): date = pd.to_datetime(date) epiweek = str(Week.fromdate(date, system="cdc")) # get epiweeks epiweek = epiweek[:4] + '_' + 'EW' + epiweek[-2:] return epiweek # add state code us_state_abbrev = { 'Alabama': 'AL', 'Alaska': 'AK', 'American Samoa': 'AS', 'Arizona': 'AZ', 'Arkansas': 'AR', 'California': 'CA', 'Colorado': 'CO', 'Connecticut': 'CT', 'Delaware': 'DE', 'District of Columbia': 'DC', 'Washington DC': 'DC', 'Florida': 'FL', 'Georgia': 'GA', 'Guam': 'GU', 'Hawaii': 'HI', 'Idaho': 'ID', 'Illinois': 'IL', 'Indiana': 'IN', 'Iowa': 'IA', 'Kansas': 'KS', 'Kentucky': 'KY', 'Louisiana': 'LA', 'Maine': 'ME', 'Maryland': 'MD', 'Massachusetts': 'MA', 'Michigan': 'MI', 'Minnesota': 'MN', 'Mississippi': 'MS', 'Missouri': 'MO', 'Montana': 'MT', 'Nebraska': 'NE', 'Nevada': 'NV', 'New Hampshire': 'NH', 'New Jersey': 'NJ', 'New Mexico': 'NM', 'New York': 'NY', 'North Carolina': 'NC', 'North Dakota': 'ND', 'Northern Mariana Islands': 'MP', 'Ohio': 'OH', 'Oklahoma': 'OK', 'Oregon': 'OR', 'Pennsylvania': 'PA', 'Puerto Rico': 'PR', 'Rhode Island': 'RI', 'South Carolina': 'SC', 'South Dakota': 'SD', 'Tennessee': 'TN', 'Texas': 'TX', 'Utah': 'UT', 'Vermont': 'VT', 'Virgin Islands': 'VI', 'Virginia': 'VA', 'Washington': 'WA', 'West Virginia': 'WV', 'Wisconsin': 'WI', 'Wyoming': 'WY' } # nextstrain metadata dfN = pd.read_csv(metadata1, encoding='utf-8', sep='\t', dtype='str') dfN['strain'] = dfN['strain'].replace('hCoV-19/', '') dfN.insert(4, 'iso', '') dfN.insert(1, 'category', '') dfN.fillna('', inplace=True) # add tag of variant category def variant_category(lineage): var_category = 'Other variants' for name in variants.keys(): if lineage == name: var_category = variants[lineage] return var_category dfN['category'] = dfN['pango_lineage'].apply(lambda x: variant_category(x)) list_columns = dfN.columns.values # list of column in the original metadata file # print(dfN) # Lab genomes metadata dfE = pd.read_excel(metadata2, index_col=None, header=0, sheet_name=0, converters={'Sample-ID': str, 'Collection-date': str}) dfE.fillna('', inplace=True) dfE = dfE.rename( columns={'Sample-ID': 'id', 'Collection-date': 'date', 'Country': 'country', 'Division (state)': 'division', 'Location (county)': 'location', 'Country of exposure': 'country_exposure', 'State of exposure': 'division_exposure', 'Lineage': 'pango_lineage', 'Source': 'originating_lab', 'Filter': 'filter'}) dfE['epiweek'] = '' # exclude rows with no ID if 'id' in dfE.columns.to_list(): dfE = dfE[~dfE['id'].isin([''])] lab_sequences = dfE['id'].tolist() # exclude unwanted lab metadata row if len(filterby) > 0: print('\nFiltering metadata by category: ' + ', '.join(filterby) + '\n') dfL = pd.DataFrame(columns=dfE.columns.to_list()) for value in filterby: dfF = dfE[dfE['filter'].isin([value])] # batch inclusion of specific rows dfL = pd.concat([dfL, dfF]) # add filtered rows to dataframe with lab metadata # list of relevant genomes sequenced keep_only = dfL['id'].tolist() excluded = [id for id in lab_sequences if id not in keep_only] # create a dict of existing sequences sequences = {} for fasta in SeqIO.parse(open(genomes), 'fasta'): # as fasta: id, seq = fasta.description, fasta.seq if id not in sequences.keys() and id not in excluded: sequences[id] = str(seq) # add inexistent columns for col in list_columns: if col not in dfL.columns: dfL[col] = '' # output dataframe outputDF = pd.DataFrame(columns=list_columns) found = [] lab_label = {} metadata_issues = {} # process metadata from excel sheet for idx, row in dfL.iterrows(): id = dfL.loc[idx, 'id'].replace('hCoV-19/', '') if id in sequences: dict_row = {} for col in list_columns: dict_row[col] = '' if col in row: dict_row[col] = dfL.loc[idx, col].strip() # add values to dictionary # check for missing geodata geodata = ['country'] # column for level in geodata: if len(dict_row[level]) < 1: if id not in metadata_issues: metadata_issues[id] = [level] else: metadata_issues[id].append(level) if dict_row['location'] in ['', None]: dict_row['location'] = dfL.loc[idx, 'location'] collection_date = '' if len(str(dict_row['date'])) > 1 and 'X' not in dict_row['date']: collection_date = dict_row['date'].split(' ')[0].replace('.', '-').replace('/', '-') dict_row['date'] = collection_date # check is date is appropriate: not from the 'future', not older than 'min_date' if pd.to_datetime(today) < pd.to_datetime(collection_date) or pd.to_datetime(min_date) > pd.to_datetime(collection_date): if id not in metadata_issues: metadata_issues[id] = ['date'] else: metadata_issues[id].append('date') else: # missing date if id not in metadata_issues: metadata_issues[id] = ['date'] else: metadata_issues[id].append('date') # fix exposure columns_exposure = ['country_exposure', 'division_exposure'] for level_exposure in columns_exposure: level = level_exposure.split('_')[0] dict_row[level_exposure] = dfL.loc[idx, level_exposure] if dict_row[level_exposure] in ['', None]: if level_exposure == 'country_exposure': dict_row[level_exposure] = dict_row[level] else: if dict_row['country_exposure'] != dfL.loc[idx, 'country']: dict_row[level_exposure] = dict_row['country_exposure'] else: dict_row[level_exposure] = dict_row[level] code = '' if dict_row['division'] in us_state_abbrev: code = us_state_abbrev[dict_row['division']] + '-' strain = dfL.loc[idx, 'country'].replace(' ', '') + '/' + code + dfL.loc[idx, 'id'] + '/' + collection_date.split('-')[0] # set the strain name dict_row['strain'] = strain dict_row['iso'] = get_iso(dict_row['country']) dict_row['originating_lab'] = dfL.loc[idx, 'originating_lab'] dict_row['submitting_lab'] = 'Grubaugh Lab - Yale School of Public Health' dict_row['authors'] = 'GLab team' # add lineage lineage = '' if dfL.loc[idx, 'pango_lineage'] != '': lineage = dfL.loc[idx, 'pango_lineage'] dict_row['pango_lineage'] = lineage # variant classication (VOI, VOC, VHC) dict_row['category'] = variant_category(lineage) # assign epiweek if len(dict_row['date']) > 0 and 'X' not in dict_row['date']: dict_row['epiweek'] = get_epiweeks(collection_date) else: dict_row['epiweek'] = '' # record sequence and metadata as found found.append(strain) if id not in metadata_issues.keys(): lab_label[id] = strain outputDF = outputDF.append(dict_row, ignore_index=True) # process metadata from TSV dfN = dfN[dfN['strain'].isin(sequences.keys())] for idx, row in dfN.iterrows(): strain = dfN.loc[idx, 'strain'].replace('hCoV-19/', '') if strain in sequences: if strain in outputDF['strain'].to_list(): continue dict_row = {} date = '' for col in list_columns: if col == 'date': date = dfN.loc[idx, col] dict_row[col] = '' if col in row: dict_row[col] = dfN.loc[idx, col] # fix exposure columns_exposure = ['country_exposure', 'division_exposure'] for level_exposure in columns_exposure: level = level_exposure.split('_')[0] if dict_row[level_exposure] in ['', None]: dict_row[level_exposure] = dict_row[level] dict_row['iso'] = get_iso(dict_row['country']) dict_row['epiweek'] = get_epiweeks(date) found.append(strain) outputDF = outputDF.append(dict_row, ignore_index=True) # write new metadata files outputDF = outputDF.drop(columns=['region']) outputDF.to_csv(output1, sep='\t', index=False) # write sequence file exported = [] with open(output2, 'w') as outfile2: # export new metadata lines for id, sequence in sequences.items(): if id in lab_label and id not in metadata_issues.keys(): # export lab generated sequences if lab_label[id] not in exported: entry = '>' + lab_label[id] + '\n' + sequence + '\n' outfile2.write(entry) print('* Exporting newly sequenced genome and metadata for ' + id) exported.append(lab_label[id]) else: # export publicly available sequences if id not in exported and id in outputDF['strain'].tolist(): entry = '>' + id + '\n' + sequence + '\n' outfile2.write(entry) exported.append(id) if len(metadata_issues) > 0: print('\n\n### WARNINGS!\n') print('\nPlease check for metadata issues related to these samples and column (which will be otherwise ignored)\n') for id, columns in metadata_issues.items(): print('\t- ' + id + ' (issues found at: ' + ', '.join(columns) + ')') print('\nMetadata file successfully reformatted and exported!\n') |
9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 | import pandas as pd from geopy.geocoders import Nominatim import argparse from bs4 import BeautifulSoup as BS import numpy as np geolocator = Nominatim(user_agent="[email protected]") # add your email here if __name__ == '__main__': parser = argparse.ArgumentParser( description="Generate file with latitudes and longitudes of samples listed in a metadata file", formatter_class=argparse.ArgumentDefaultsHelpFormatter ) parser.add_argument("--metadata", required=True, help="Nextstrain metadata file") parser.add_argument("--geoscheme", required=True, help="XML file with geographic schemes") parser.add_argument("--columns", nargs='+', type=str, help="list of columns that need coordinates") parser.add_argument("--cache", required=False, help="TSV file with preexisting latitudes and longitudes") parser.add_argument("--output", required=True, help="TSV file containing geographic coordinates") args = parser.parse_args() metadata = args.metadata geoscheme = args.geoscheme columns = args.columns cache = args.cache output = args.output # metadata = path + 'metadata_geo.tsv' # geoscheme = path + "geoscheme.tsv" # columns = ['region', 'country', 'division', 'location'] # cache = path + 'cache_coordinates.tsv' # output = path + 'latlongs.tsv' force_coordinates = {'Washington DC': ('38.912708', '-77.009223'), 'New-York-State': ('43.1561681', '-75.8449946'), 'Puerto Rico': ('18.235853', '-66.522056'), 'Virgin Islands': ('17.727304', '-64.748327'), 'Indiana': ('39.768534', '-86.158011'), 'Middlesex':('41.482782', '-72.556658')} results = {trait: {} for trait in columns} # content to be exported as final result # extract coordinates from cache file try: for line in open(cache).readlines(): if not line.startswith('\n'): try: trait, place, lat, long = line.strip().split('\t') if trait in results.keys(): entry = {place: (str(lat), str(long))} results[trait].update(entry) # save as pre-existing result except: pass except: pass # extract coordinates from TSV file scheme_list = open(geoscheme, "r").readlines()[1:] dont_search = [] set_countries = [] for line in scheme_list: if not line.startswith('\n'): type = line.split('\t')[0] if type in columns: coordinates = {} try: subarea = line.split('\t')[2] # name of the pre-defined area in the TSV file lat = line.split('\t')[3] long = line.split('\t')[4] entry = {subarea: (lat, long)} coordinates.update(entry) if subarea not in results[type]: results[type].update(coordinates) dont_search.append(subarea) country_name = subarea.split('-')[0] if type == 'country' and country_name not in set_countries: set_countries.append(country_name) except: pass # find coordinates for locations not found in cache or XML file def find_coordinates(place): try: location = geolocator.geocode(place, language='en') lat, long = location.latitude, location.longitude coord = (str(lat), str(long)) return coord except: coord = ('NA', 'NA') return coord # open metadata file as dataframe dfN = pd.read_csv(metadata, encoding='utf-8', sep='\t') queries = [] pinpoints = [dfN[trait].values.tolist() for trait in columns if trait != 'region'] for address in zip(*pinpoints): traits = [trait for trait in columns if trait != 'region'] for position, place in enumerate(address): level = traits[position] query = list(address[0:position + 1]) queries.append((level, query)) not_found = [] for unknown_place in queries: trait, place = unknown_place[0], unknown_place[1] target = place[-1] if target not in ['', 'NA', 'NAN', 'unknown', '-', np.nan, None]: try: if place[0].split('-')[0] in set_countries: country_short = place[0].split('-')[0] # correcting TSV pre-defined country names place[0] = country_short except: pass if target not in results[trait]: new_query = [] for name in place: if name not in dont_search: if place[0] == 'USA': if name != 'USA': name = name + ' state' if name not in new_query: new_query.append(name) item = (trait, ', '.join(new_query)) coord = ('NA', 'NA') if item not in not_found: coord = find_coordinates(', '.join(new_query)) # search coordinates if 'NA' in coord: if item not in not_found: not_found.append(item) print('\t* WARNING! Coordinates not found for: ' + trait + ', ' + ', '.join(new_query)) else: print(trait + ', ' + target + '. Coordinates = ' + ', '.join(coord)) entry = {target: coord} results[trait].update(entry) print('\n### These coordinates were found and saved in the output file:') with open(output, 'w') as outfile: for trait, lines in results.items(): print('\n* ' + trait) for place, coord in lines.items(): if place in force_coordinates: lat, long = force_coordinates[place][0], force_coordinates[place][1] else: lat, long = coord[0], coord[1] print(place + ': ' + lat + ', ' + long) line = "{}\t{}\t{}\t{}\n".format(trait, place, lat, long) outfile.write(line) outfile.write('\n') if len(not_found) > 1: print('\n### WARNING! Some coordinates were not found (see below).' '\nTypos or especial characters in place names my explain such errors.' '\nPlease fix them, and run the script again, or add coordinates manually:\n') for trait, address in not_found: print(trait + ': ' + address) print('\nCoordinates file successfully created!\n') |
6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 | import argparse import Bio import Bio.SeqIO from Bio.Seq import Seq if __name__ == '__main__': parser = argparse.ArgumentParser( description="Mask initial bases from alignment FASTA", formatter_class=argparse.ArgumentDefaultsHelpFormatter ) parser.add_argument("--alignment", required=True, help="FASTA file of alignment") parser.add_argument("--mask-from-beginning", type = int, required=True, help="number of bases to mask from start") parser.add_argument("--mask-from-end", type = int, help="number of bases to mask from end") parser.add_argument("--mask-sites", nargs='+', type = int, help="list of sites to mask") parser.add_argument("--output", required=True, help="FASTA file of output alignment") args = parser.parse_args() being_length = 0 if args.mask_from_beginning: begin_length = args.mask_from_beginning end_length = 0 if args.mask_from_end: end_length = args.mask_from_end with open(args.output, 'w') as outfile: for record in Bio.SeqIO.parse(args.alignment, 'fasta'): seq = str(record.seq) start = "N" * begin_length middle = seq[begin_length:-end_length] end = "N" * end_length seq_list = list(start + middle + end) if args.mask_sites: for site in args.mask_sites: seq_list[site-1] = "N" record.seq = Seq("".join(seq_list)) Bio.SeqIO.write(record, outfile, 'fasta') |
9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 | import pandas as pd import argparse if __name__ == '__main__': parser = argparse.ArgumentParser( description="Merge two excel spreadsheets and export .xlxs file", formatter_class=argparse.ArgumentDefaultsHelpFormatter ) parser.add_argument("--sheet1", required=True, help="Spreadsheet file 1") parser.add_argument("--sheet2", required=True, help="Spreadsheet file 2") parser.add_argument("--index", required=True, default='Sample-ID', type=str, help="Name of column with unique identifiers") parser.add_argument("--output", required=True, help="Merged spreadsheet file") args = parser.parse_args() sheet1 = args.sheet1 sheet2 = args.sheet2 index = args.index output = args.output # sheet1 = path + 'COVID-19_sequencing.xlsx' # sheet2 = path + 'Incoming S drop out samples.xlsx' # index = 'Sample-ID' # output = path + 'merged_sheet.xlsx' # load spreadsheets df1 = pd.read_excel(sheet1, index_col=None, header=0, sheet_name=0) df1.fillna('', inplace=True) df1 = df1[~df1[index].isin([''])] # drop row with empty index df2 = pd.read_excel(sheet2, index_col=None, header=0, sheet_name=0) df2.fillna('', inplace=True) df2 = df2[~df2[index].isin([''])] # drop row with empty index # empty matrix dataframe columns = [x for x in df1.columns.to_list() if x in df2.columns.to_list()] rows = [x for x in df1[index].to_list() if x not in df2[index].to_list()] + df2[index].to_list() duplicates = [x for x in df1[index].to_list() if x in df2[index].to_list()] # drop duplicates df2 = df2[~df2[index].isin(duplicates)] # filter columns df1 = df1[columns] df2 = df2[columns] # filter rows df1 = df1[df1[index].isin(rows)] df2 = df2[df2[index].isin(rows)] # concatenate dataframes frames = [df1, df2] df3 = pd.concat(frames) df3 = df3.astype(str) # export Excel sheet df3.to_excel(output, index=False) print('\nSpreadsheets successfully merged.\n') |
57 58 59 60 61 62 63 64 65 | shell: """ python3 scripts/add_genomes.py \ --genomes {input.genomes} \ --new-genomes {input.new_genomes} \ --keep {input.include} \ --remove {input.exclude} \ --output {output.sequences} """ |
80 81 82 83 84 85 86 87 | shell: """ python3 scripts/merge_sheets.py \ --sheet1 {input.metadata1} \ --sheet2 {input.metadata2} \ --index {params.index} \ --output {output.merged_metadata} \ """ |
107 108 109 110 111 112 113 114 115 116 | shell: """ python3 scripts/filter_metadata.py \ --genomes {input.genomes} \ --metadata1 {input.metadata1} \ --metadata2 {input.metadata2} \ --filter {params.filter} \ --output1 {output.filtered_metadata} \ --output2 {output.sequences} """ |
132 133 134 135 136 137 138 139 | shell: """ python3 scripts/apply_geoscheme.py \ --metadata {input.filtered_metadata} \ --geoscheme {input.geoscheme} \ --output {output.final_metadata} \ --filter {params.filter} """ |
155 156 157 158 159 160 161 162 163 164 | shell: """ python3 scripts/get_coordinates.py \ --metadata {input.metadata} \ --geoscheme {input.geoscheme} \ --columns {params.columns} \ --cache {input.cache} \ --output {output.latlongs} cp {output.latlongs} config/cache_coordinates.tsv """ |
182 183 184 185 186 187 188 189 190 191 192 | shell: """ python3 scripts/apply_colour_scheme.py \ --metadata {input.metadata} \ --coordinates {input.latlongs} \ --geoscheme {input.geoscheme} \ --grid {input.colour_grid} \ --columns {params.columns} \ --output {output.colours} \ --filter {params.filt} """ |
223 224 225 226 227 228 229 230 | shell: """ augur filter \ --sequences {input.sequences} \ --metadata {input.metadata} \ --exclude {input.exclude} \ --output {output.sequences} """ |
248 249 250 251 252 253 254 255 256 257 | shell: """ augur align \ --sequences {input.sequences} \ --existing-alignment {files.aligned} \ --reference-sequence {input.reference} \ --nthreads {params.threads} \ --output {output.alignment} \ --remove-reference """ |
277 278 279 280 281 282 283 284 285 | shell: """ python3 scripts/mask-alignment.py \ --alignment {input.alignment} \ --mask-from-beginning {params.mask_from_beginning} \ --mask-from-end {params.mask_from_end} \ --mask-sites {params.mask_sites} \ --output {output.alignment} """ |
298 299 300 301 302 303 304 | shell: """ augur tree \ --alignment {input.alignment} \ --nthreads {params.threads} \ --output {output.tree} """ |
331 332 333 334 335 336 337 338 339 340 341 342 343 344 345 346 347 348 | shell: """ augur refine \ --tree {input.tree} \ --alignment {input.alignment} \ --metadata {input.metadata} \ --output-tree {output.tree} \ --output-node-data {output.node_data} \ --root {params.root} \ --timetree \ --coalescent {params.coalescent} \ --date-confidence \ --clock-filter 4 \ --clock-rate {params.clock_rate} \ --clock-std-dev {params.clock_std_dev} \ --divergence-units {params.unit} \ --date-inference {params.date_inference} """ |
363 364 365 366 367 368 369 370 | shell: """ augur ancestral \ --tree {input.tree} \ --alignment {input.alignment} \ --inference {params.inference} \ --output-node-data {output.node_data} """ |
382 383 384 385 386 387 388 389 | shell: """ augur translate \ --tree {input.tree} \ --ancestral-sequences {input.node_data} \ --reference-sequence {input.reference} \ --output {output.node_data} \ """ |
403 404 405 406 407 408 409 410 411 | shell: """ augur traits \ --tree {input.tree} \ --metadata {input.metadata} \ --output {output.node_data} \ --columns {params.columns} \ --confidence """ |
425 426 427 428 429 430 431 | shell: """ augur clades --tree {input.tree} \ --mutations {input.nuc_muts} {input.aa_muts} \ --clades {input.clades} \ --output {output.clade_data} """ |
450 451 452 453 454 455 456 457 458 459 460 461 462 | shell: """ augur frequencies \ --method kde \ --metadata {input.metadata} \ --tree {input.tree} \ --min-date {params.min_date} \ --pivot-interval {params.pivot_interval} \ --pivot-interval-units {params.pivot_interval_units} \ --narrow-bandwidth {params.narrow_bandwidth} \ --proportion-wide {params.proportion_wide} \ --output {output.tip_frequencies_json} 2>&1 | tee {log} """ |
482 483 484 485 486 487 488 489 490 491 492 | shell: """ augur export v2 \ --tree {input.tree} \ --metadata {input.metadata} \ --node-data {input.branch_lengths} {input.traits} {input.nt_muts} {input.aa_muts} {input.clades} \ --colors {input.colors} \ --lat-longs {input.lat_longs} \ --auspice-config {input.auspice_config} \ --output {output.auspice} """ |
504 505 506 507 | shell: """ rm -rfv {params} """ |
521 522 523 524 | shell: """ rm -rfv {params} """ |
532 533 534 535 | shell: """ rm -rfv {params} """ |
542 543 | shell: "rm -rfv {params}" |
Support
- Share:
-

-

-

- Future updates
Related Workflows





