Python Mutation Free Energy Calculations using BioExcel Building Blocks (biobb)





Help improve this workflow!
This workflow has been published but could be further improved with some additional meta data:- Keyword(s) in categories input, output, operation
You can help improve this workflow by suggesting the addition or removal of keywords, suggest changes and report issues, or request to become a maintainer of the Workflow .
Mutation Free Energy Calculations using BioExcel Building Blocks (biobb)
Based on the official pmx tutorial .
This tutorial aims to illustrate how to compute a fast-growth mutation free energy calculation, step by step, using the BioExcel Building Blocks library (biobb) . The particular example used is the Staphylococcal nuclease protein (PDB code 1STN), a small, minimal protein, appropriate for a short tutorial.
The non-equilibrium free energy calculation protocol performs a fast alchemical transition in the direction WT->Mut and back Mut->WT . The two equilibrium trajectories needed for the tutorial, one for Wild Type (WT) and another for the Mutated (Mut) protein (Isoleucine 10 to Alanine -I10A-), have already been generated and are included in this example. We will name WT as stateA and Mut as stateB .
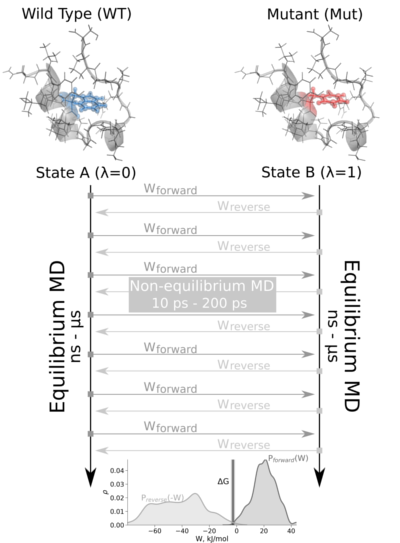
The tutorial calculates the free energy difference in the folded state of a protein. Starting from two 1ns-length independent equilibrium simulations (WT and mutant), snapshots are selected to start fast (50ps) transitions driving the system in the forward (WT to mutant) and reverse (mutant to WT) directions, and the work values required to perform these transitions are collected. With these values, Crooks Gaussian Intersection (CGI), Bennett Acceptance Ratio (BAR) and Jarzynski estimator methods are used to calculate the free energy difference between the two states.
![]()
Code Snippets
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 | import time import argparse import os import zipfile # biobb common modules from biobb_common.configuration import settings from biobb_common.tools import file_utils as fu # biobb pmx modules from biobb_pmx.pmxbiobb.pmxmutate import pmxmutate from biobb_pmx.pmxbiobb.pmxgentop import pmxgentop from biobb_pmx.pmxbiobb.pmxanalyse import pmxanalyse # biobb md modules from biobb_gromacs.gromacs.pdb2gmx import pdb2gmx from biobb_gromacs.gromacs.make_ndx import make_ndx from biobb_gromacs.gromacs.grompp import grompp from biobb_gromacs.gromacs.mdrun import mdrun # biobb analysis module from biobb_analysis.gromacs.gmx_trjconv_str_ens import gmx_trjconv_str_ens def main(config, system=None): start_time = time.time() conf = settings.ConfReader(config, system) global_log, _ = fu.get_logs(path=conf.get_working_dir_path(), light_format=True) global_prop = conf.get_prop_dic(global_log=global_log) global_paths = conf.get_paths_dic() dhdl_paths_listA = [] dhdl_paths_listB = [] for ensemble, mutation in conf.properties['mutations'].items(): ensemble_prop = conf.get_prop_dic(prefix=ensemble, global_log=global_log) ensemble_paths = conf.get_paths_dic(prefix=ensemble) # Create and launch bb global_log.info(ensemble+" Step 0: gmx trjconv: Extract snapshots from equilibrium trajectories") ensemble_paths['step0_trjconv']['input_traj_path'] = conf.properties['input_trajs'][ensemble]['input_traj_path'] ensemble_paths['step0_trjconv']['input_top_path'] = conf.properties['input_trajs'][ensemble]['input_tpr_path'] gmx_trjconv_str_ens(**ensemble_paths["step0_trjconv"], properties=ensemble_prop["step0_trjconv"]) with zipfile.ZipFile(ensemble_paths["step0_trjconv"]["output_str_ens_path"], 'r') as zip_f: zip_f.extractall() state_pdb_list = zip_f.namelist() for pdb_path in state_pdb_list: pdb_name = os.path.splitext(pdb_path)[0] prop = conf.get_prop_dic(prefix=os.path.join(ensemble, pdb_name), global_log=global_log) paths = conf.get_paths_dic(prefix=os.path.join(ensemble, pdb_name)) # Create and launch bb global_log.info("Step 1: pmx mutate: Generate Hybrid Structure") paths['step1_pmx_mutate']['input_structure_path'] = pdb_path prop['step1_pmx_mutate']['mutation_list'] = mutation pmxmutate(**paths["step1_pmx_mutate"], properties=prop["step1_pmx_mutate"]) # Step 2: gmx pdb2gmx: Generate Topology # From pmx tutorial: # gmx pdb2gmx -f mut.pdb -ff amber99sb-star-ildn-mut -water tip3p -o pdb2gmx.pdb global_log.info("Step 2: gmx pdb2gmx: Generate Topology") pdb2gmx(**paths["step2_gmx_pdb2gmx"], properties=prop["step2_gmx_pdb2gmx"]) # Step 3: pmx gentop: Generate Hybrid Topology # From pmx tutorial: # python generate_hybrid_topology.py -itp topol_Protein.itp -o topol_Protein.itp -ff amber99sb-star-ildn-mut global_log.info("Step 3: pmx gentop: Generate Hybrid Topology") pmxgentop(**paths["step3_pmx_gentop"], properties=prop["step3_pmx_gentop"]) # Step 4: gmx make_ndx: Generate Gromacs Index File to select atoms to freeze # From pmx tutorial: # echo -e "a D*\n0 & ! 19\nname 20 FREEZE\nq\n" | gmx make_ndx -f frame0/pdb2gmx.pdb -o index.ndx global_log.info("Step 4: gmx make_ndx: Generate Gromacs Index file to select atoms to freeze") make_ndx(**paths["step4_gmx_makendx"], properties=prop["step4_gmx_makendx"]) if ensemble == 'stateA': # In stateA, with lamdda=0, we don't need the energy minimization step, so simply get the output # from the step2 (pdb2gmx) as output from the step6 (energy minimization) # From pmx tutorial: # There are no dummies in this state at lambda=0, therefore simply convert mut.pdb to emout.gro paths['step7_gmx_grompp']['input_gro_path'] = paths['step2_gmx_pdb2gmx']['output_gro_path'] elif ensemble == 'stateB': # Step 5: gmx grompp: Creating portable binary run file for energy minimization # From pmx tutorial: # gmx grompp -c pdb2gmx.pdb -p topol.top -f ../../mdp/em_FREEZE.mdp -o em.tpr -n ../index.ndx global_log.info("Step 5: gmx grompp: Creating portable binary run file for energy minimization") grompp(**paths["step5_gmx_grompp"], properties=prop["step5_gmx_grompp"]) # Step 6: gmx mdrun: Running energy minimization # From pmx tutorial: # gmx mdrun -s em.tpr -c emout.gro -v global_log.info(ensemble+" Step 6: gmx mdrun: Running energy minimization") mdrun(**paths["step6_gmx_mdrun"], properties=prop["step6_gmx_mdrun"]) # Step 7: gmx grompp: Creating portable binary run file for system equilibration # From pmx tutorial: # gmx grompp -c emout.gro -p topol.top -f ../../mdp/eq_20ps.mdp -o eq_20ps.tpr -maxwarn 1 global_log.info(ensemble+" Step 7: gmx grompp: Creating portable binary run file for system equilibration") grompp(**paths["step7_gmx_grompp"], properties=prop["step7_gmx_grompp"]) # Step 8: gmx mdrun: Running system equilibration # From pmx tutorial: # gmx mdrun -s eq_20ps.tpr -c eqout.gro -v global_log.info(ensemble+" Step 8: gmx mdrun: Running system equilibration") mdrun(**paths["step8_gmx_mdrun"], properties=prop["step8_gmx_mdrun"]) # Step 9: gmx grompp: Creating portable binary run file for thermodynamic integration (ti) # From pmx tutorial: # gmx grompp -c eqout.gro -p topol.top -f ../../mdp/ti.mdp -o ti.tpr -maxwarn 1 global_log.info(ensemble+" Step 9: Creating portable binary run file for thermodynamic integration (ti)") grompp(**paths["step9_gmx_grompp"], properties=prop["step9_gmx_grompp"]) # Step 10: gmx mdrun: Running thermodynamic integration # From pmx tutorial: # gmx mdrun -s ti.tpr -c eqout.gro -v global_log.info(ensemble+" Step 10: gmx mdrun: Running thermodynamic integration") mdrun(**paths["step10_gmx_mdrun"], properties=prop["step10_gmx_mdrun"]) if ensemble == "stateA": dhdl_paths_listA.append(paths["step10_gmx_mdrun"]["output_dhdl_path"]) elif ensemble == "stateB": dhdl_paths_listB.append(paths["step10_gmx_mdrun"]["output_dhdl_path"]) # Creating zip file containing all the dhdl files dhdlA_path = 'dhdlA.zip' dhdlB_path = 'dhdlB.zip' fu.zip_list(dhdlA_path, dhdl_paths_listA, global_log) fu.zip_list(dhdlB_path, dhdl_paths_listB, global_log) # Step 11: pmx analyse: Calculate free energies from fast growth thermodynamic integration simulations # From pmx tutorial: # python analyze_dhdl.py -fA ../stateA/frame*/dhdl*.xvg -fB ../stateB/frame*/dhdl*.xvg --nbins 25 -t 293 --reverseB global_log.info(ensemble+" Step 11: pmx analyse: Calculate free energies from fast growth thermodynamic integration simulations") global_paths["step11_pmx_analyse"]["input_a_xvg_zip_path"] = dhdlA_path global_paths["step11_pmx_analyse"]["input_b_xvg_zip_path"] = dhdlB_path pmxanalyse(**global_paths["step11_pmx_analyse"], properties=global_prop["step11_pmx_analyse"]) elapsed_time = time.time() - start_time global_log.info('') global_log.info('') global_log.info('Execution successful: ') global_log.info(' Workflow_path: %s' % conf.get_working_dir_path()) global_log.info(' Config File: %s' % config) if system: global_log.info(' System: %s' % system) global_log.info('') global_log.info('Elapsed time: %.1f minutes' % (elapsed_time/60)) global_log.info('') if __name__ == '__main__': parser = argparse.ArgumentParser(description="Automatic Ligand parameterization tutorial using BioExcel Building Blocks") parser.add_argument('--config', required=True) parser.add_argument('--system', required=False) args = parser.parse_args() main(args.config, args.system) |
Support
- Share:
-

-

-

- Future updates
Related Workflows





