




Rare disease researchers workflow is that they submit their raw data (fastq), run the mapping and variant calling RD-Connect pipeline and obtain unannotated gvcf files to further submit to the RD-Connect GPAP or analyse on their own.
This demonstrator focuses on the variant calling pipeline. The raw genomic data is processed using the RD-Connect pipeline ( Laurie et al., 2016 ) running on the standards (GA4GH) compliant, interoperable container orchestration platform.
This demonstrator will be aligned with the current implementation study on Development of Architecture for Software Containers at ELIXIR and its use by EXCELERATE use-case communities
For this implementation, different steps are required:
- Adapt the pipeline to CWL and dockerise elements
- Align with IS efforts on software containers to package the different components (Nextflow)
- Submit trio of Illumina NA12878 Platinum Genome or Exome to the GA4GH platform cloud (by Aspera or ftp server)
- Run the RD-Connect pipeline on the container platform
- Return corresponding gvcf files
- OPTIONAL: annotate and update to RD-Connect playground instance
N.B: The demonstrator might have some manual steps, which will not be in production.
RD-Connect pipeline
Detailed information about the RD-Connect pipeline can be found in Laurie et al., 2016
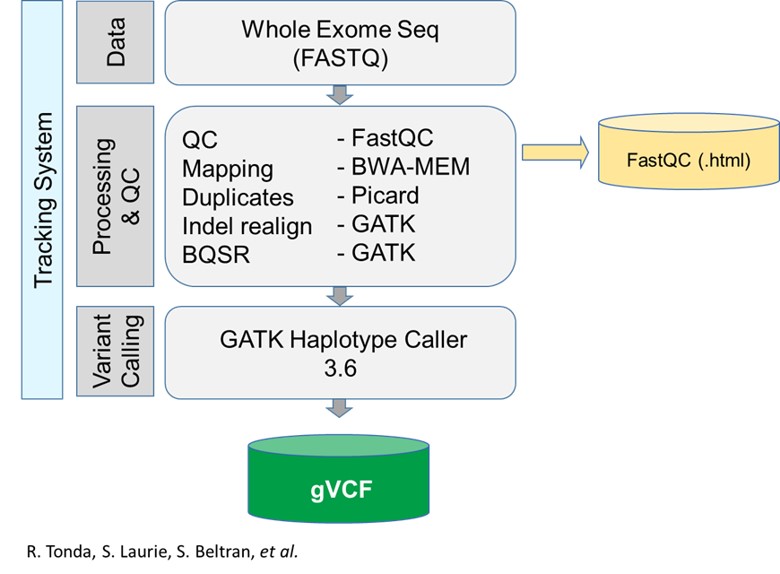
The applications
1. Name of the application: Adaptor removal
Function: remove sequencing adaptors
Container (readiness status, location, version):
cutadapt (v.1.18)
Required resources in cores and RAM: current container size 169MB
Input data (amount, format, directory..): raw fastq
Output data: paired fastq without adaptors
2. Name of the application: Mapping and bam sorting
Function: align data to reference genome
Container :
bwa-mem (v.0.7.17)
/
Sambamba (v. 0.6.8 )
(or samtools)
Resources :current container size 111MB / 32MB
Input data: paired fastq without adaptors
Output data: sorted bam
3. Name of the application: MarkDuplicates
Function: Mark (and remove) duplicates
Container:
Picard (v.2.18.25)
Resources: current container size 261MB
Input data:sorted bam
Output data: Sorted bam with marked (or removed) duplicates
4. Name of the application: Base quality recalibration (BQSR)
Function: Base quality recalibration
Container:
GATK (v.3.6-0)
Resources: current container size 270MB
Input data: Sorted bam with marked (or removed) duplicates
Output data: Sorted bam with marked duplicates & base quality recalculated
5. Name of the application: Variant calling
Function: variant calling
Container:
GATK (v.3.6-0)
Resources: current container size 270MB
Input data:Sorted bam with marked duplicates & base quality recalculated
Output data: unannotated gvcf per sample
6. (OPTIONAL)Name of the application: Quality of the fastq
Function: report on the sequencing quality
Container:
fastqc 0.11.8
Resources: current container size 173MB
Input data: raw fastq
Output data: QC report
Licensing
GATK declares that archived packages are made available for free to academic researchers under a limited license for non-commercial use. If you need to use one of these packages for commercial use. https://software.broadinstitute.org/gatk/download/archive
Code Snippets
269 270 271 272 273 274 275 276 277 | """ cutadapt \ ${adaptorArgument} \ ${cutadaptcoresArgument} \ ${params.cutadapt.extraargs} \ --output ${read1.baseName}.noAdaptor.fastq.gz \ --paired-output ${read2.baseName}.noAdaptor.fastq.gz \ $read1 $read2 """ |
334 335 336 | """ bwa index -a bwtsw ${bwablocksize} $reference """ |
372 373 374 | """ bwa mem -R "${params.bwamem.rgheader}" -M ${bwacoresArgument} ${reference.name} ${read1} ${read2} > ${reference.name}.sam """ |
393 394 395 396 397 398 399 | """ sambamba view \ ${sambambacoresArgument} \ -S ${reference.name}.sam \ -o ${reference.name}.bam \ -f bam """ |
422 423 424 425 426 427 428 | """ sambamba sort \ ${sambambamemorylevelhint} \ ${sambambaCompressionArgument} \ ${reference.name}.bam \ > ${reference.name}.sorted.bam """ |
486 487 488 | """ picard MarkDuplicates ${picardCMD} """ |
501 502 503 | """ gunzip -c ${reference} > ${reference.baseName} """ |
515 516 517 | """ samtools faidx ${reference.baseName} """ |
529 530 531 | """ picard CreateSequenceDictionary REFERENCE=${reference.baseName} OUTPUT=${reference.getBaseName(2)}.dict """ |
543 544 545 | """ samtools index ${reference.name}.sorted.noDuplicates.bam """ |
583 584 585 586 587 588 589 590 591 592 593 594 595 596 597 598 599 600 601 602 603 604 605 606 607 608 609 610 | """ #!/bin/bash mkdir -p knownSites for vcf in knownSitesInput/* ; do case "\${vcf}" in *.gz) gunzip -c "\${vcf}" > knownSites/"\$(basename "\${vcf}" .gz)" ;; *) ln -sf ../"\${vcf}" knownSites ;; esac done knownSitesArg="" for vcf in knownSites/*.vcf ; do knownSitesArg+=" --knownSites \${vcf}" done java -jar /usr/GenomeAnalysisTK.jar \ -T BaseRecalibrator \ -nct 8 \ -R ${reference.baseName} \ --input_file ${reference.name}.sorted.noDuplicates.bam \ -dt NONE \ -o ${reference.name}.sorted.noDuplicates.recalibrated.grp \ \${knownSitesArg} """ |
628 629 630 631 632 633 634 635 | """ java -jar /usr/GenomeAnalysisTK.jar \ -T PrintReads \ -R ${reference.baseName} \ -I ${reference.name}.sorted.noDuplicates.bam \ -BQSR ${reference.name}.sorted.noDuplicates.recalibrated.grp \ -o ${reference.name}.sorted.noDuplicates.recalibrated.printed.bam """ |
662 663 664 665 666 667 668 669 670 671 672 673 674 | """ java -jar /usr/GenomeAnalysisTK.jar \ -T HaplotypeCaller \ --num_cpu_threads_per_data_thread ${params.bwahaplotyper.cputhreads} \ -I ${reference.name}.sorted.noDuplicates.recalibrated.printed.bam \ -R ${reference.baseName} \ --min_base_quality_score ${params.bwahaplotyper.minquality} \ -ERC ${params.bwahaplotyper.erc} \ -variant_index_type ${params.bwahaplotyper.variantindextype} \ --variant_index_parameter ${params.bwahaplotyper.variantindexparam} \ -o ${reference.baseName}.sorted.noDuplicates.recalibrated.g.vcf.gz \ ${GQB} """ |
Support
- Share:
-

-

-

- Future updates
Related Workflows





