detection, annotation, and taxonomic classification of viral contigs in metagenomic and metatranscriptomic assemblies





![]()
![]()

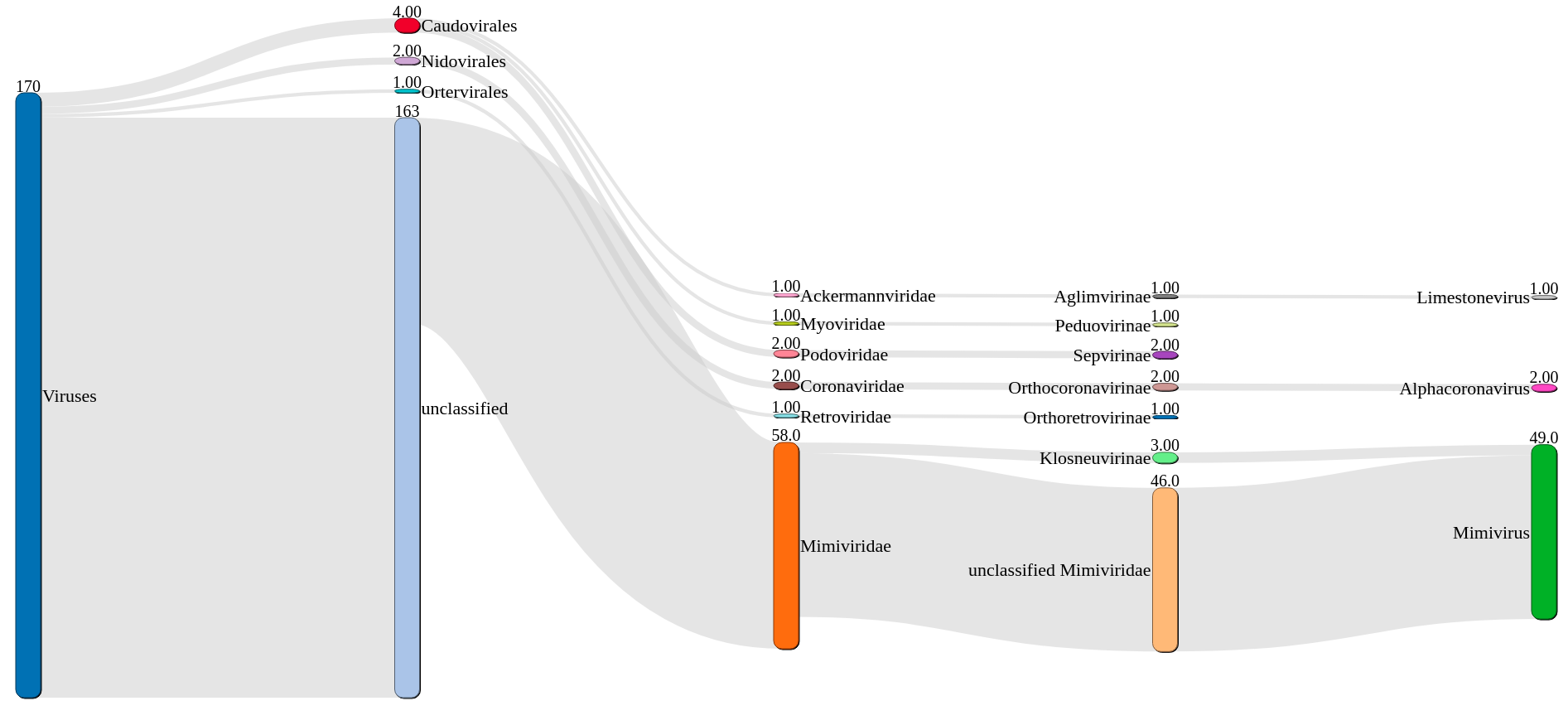
VIRify is a recently developed pipeline for the detection, annotation, and taxonomic classification of viral contigs in metagenomic and metatranscriptomic assemblies. The pipeline is part of the repertoire of analysis services offered by MGnify . VIRify’s taxonomic classification relies on the detection of taxon-specific profile hidden Markov models (HMMs), built upon a set of 22,014 orthologous protein domains and referred to as ViPhOGs.
VIRify was implemented in CWL.
What do I need?
The current implementation uses CWL version 1.2 dev+2. It was tested using Toil version 4.10 as the workflow engine and conda to manage the software dependencies.
Docker - Singularity support
Soon…
Setup environment
conda env create -f cwl/requirements/conda_env.yml
conda activate viral_pipeline
Basic execution
cd cwl/
virify.sh -h
A note about metatranscriptomes
Although VIRify has been benchmarked and validated with metagenomic data in mind, it is also possible to use this tool to detect RNA viruses in metatranscriptome assemblies (e.g. SARS-CoV-2). However, some additional considerations for this purpose are outlined below:
1. Quality control:
As for metagenomic data, a thorough quality control of the FASTQ sequence reads to remove low-quality bases, adapters and host contamination (if appropriate) is required prior to assembly. This is especially important for metatranscriptomes as small errors can further decrease the quality and contiguity of the assembly obtained. We have used
TrimGalore
for this purpose.
2. Assembly: There are many assemblers available that are appropriate for either metagenomic or single-species transcriptomic data. However, to our knowledge, there is no assembler currently available specifically for metatranscriptomic data. From our preliminary investigations, we have found that transcriptome-specific assemblers (e.g. rnaSPAdes ) generate more contiguous and complete metatranscriptome assemblies compared to metagenomic alternatives (e.g. MEGAHIT and metaSPAdes ).
3. Post-processing: Metatranscriptomes generate highly fragmented assemblies. Therefore, filtering contigs based on a set minimum length has a substantial impact in the number of contigs processed in VIRify. It has also been observed that the number of false-positive detections of VirFinder (one of the tools included in VIRify) is lower among larger contigs. The choice of a length threshold will depend on the complexity of the sample and the sequencing technology used, but in our experience any contigs <2 kb should be analysed with caution.
4. Classification: The classification module of VIRify depends on the presence of a minimum number and proportion of phylogenetically-informative genes within each contig in order to confidently assign a taxonomic lineage. Therefore, short contigs typically obtained from metatranscriptome assemblies remain generally unclassified. For targeted classification of RNA viruses (for instance, to search for Coronavirus-related sequences), alternative DNA- or protein-based classification methods can be used. Two of the possible options are: (i) using MashMap to screen the VIRify contigs against a database of RNA viruses (e.g. Coronaviridae) or (ii) using hmmsearch to screen the proteins obtained in the VIRify contigs against marker genes of the taxon of interest.
Contact us
MGnify helpdeskCode Snippets
13 14 15 | """ viral_contigs_annotation.py -o . -p ${faa} -t ${tab} """ |
15 16 17 | """ contig_taxonomic_assign.py -i ${tab} -d ${db} --factor ${factor} --taxthres ${params.taxthres} """ |
12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 | """ if [[ "${set_name}" == "all" ]]; then # concat all taxonomy results per assembly regardless of HC, LC or PP cat *_annotation_taxonomy.tsv | grep -v contig_ID > tmp.tsv NAME=all else cp ${tbl} tmp.tsv NAME=${set_name} fi # genus grep -v contig_ID tmp.tsv | awk -v SAMPLE="${name}" 'BEGIN{FS="\\t"};{if(\$2!="" && \$2 !~ /^0/){print SAMPLE"\\tgenus\\t"\$2}}' | sort | uniq -c | awk '{printf \$2"\\t"\$3"\\t"\$4"\\t"\$1"\\n"}' > \$NAME"_summary.tsv" # subfamily grep -v contig_ID tmp.tsv | awk -v SAMPLE="${name}" 'BEGIN{FS="\\t"};{if(\$3!="" && \$3 !~ /^0/){print SAMPLE"\\tsubfamily\\t"\$3}}' | sort | uniq -c | awk '{printf \$2"\\t"\$3"\\t"\$4"\\t"\$1"\\n"}' >> \$NAME"_summary.tsv" # family grep -v contig_ID tmp.tsv | awk -v SAMPLE="${name}" 'BEGIN{FS="\\t"};{if(\$4!="" && \$4 !~ /^0/){print SAMPLE"\\tfamily\\t"\$4}}' | sort | uniq -c | awk '{printf \$2"\\t"\$3"\\t"\$4"\\t"\$1"\\n"}' >> \$NAME"_summary.tsv" # order grep -v contig_ID tmp.tsv | awk -v SAMPLE="${name}" 'BEGIN{FS="\\t"};{if(\$5!="" && \$5 !~ /^0/){print SAMPLE"\\torder\\t"\$5}}' | sort | uniq -c | awk '{printf \$2"\\t"\$3"\\t"\$4"\\t"\$1"\\n"}' >> \$NAME"_summary.tsv" if [ -s \$NAME"_summary.tsv" ]; then balloon.R "\${NAME}_summary.tsv" "\${NAME}_balloon.svg" 10 8 fi """ |
18 19 20 | """ imgvr_merge.py -f ${blast_filtered} -d IMG_VR_2018-07-01_4/IMGVR_all_Sequence_information.tsv -o \$(basename ${blast_filtered} .blast).meta """ |
22 23 24 | """ imgvr_merge.py -f ${blast_filtered} -d ${db}/IMG_VR_2018-07-01_4/IMGVR_all_Sequence_information.tsv -o \$(basename ${blast_filtered} .blast).meta """ |
18 19 20 21 22 23 24 25 | """ sed -i "s/ /|/" ${fasta} HEADER_BLAST="qseqid\\tsseqid\\tpident\\tlength\\tmismatch\\tgapopen\\tqstart\\tqend\\tqlen\\tsstart\\tsend\\tevalue\\tbitscore\\tslen" printf "\$HEADER_BLAST\\n" > ${confidence_set_name}.blast blastn -task blastn -num_threads ${task.cpus} -query ${fasta} -db IMG_VR_2018-07-01_4/IMGVR_all_nucleotides.fna -evalue 1e-10 -outfmt "6 qseqid sseqid pident length mismatch gapopen qstart qend qlen sstart send evalue bitscore slen" >> ${confidence_set_name}.blast awk '{if(\$4>0.8*\$9){print \$0}}' ${confidence_set_name}.blast >> ${confidence_set_name}.filtered.blast """ |
27 28 29 30 31 32 33 34 | """ sed -i "s/ /|/" ${fasta} HEADER_BLAST="qseqid\\tsseqid\\tpident\\tlength\\tmismatch\\tgapopen\\tqstart\\tqend\\tqlen\\tsstart\\tsend\\tevalue\\tbitscore\\tslen" printf "\$HEADER_BLAST\\n" > ${confidence_set_name}.blast blastn -task blastn -num_threads ${task.cpus} -query ${fasta} -db ${db}/IMG_VR_2018-07-01_4/IMGVR_all_nucleotides.fna -evalue 1e-10 -outfmt "6 qseqid sseqid pident length mismatch gapopen qstart qend qlen sstart send evalue bitscore slen" >> ${confidence_set_name}.blast awk '{if(\$4>0.8*\$9){print \$0}}' ${confidence_set_name}.blast >> ${confidence_set_name}.filtered.blast """ |
8 9 10 11 12 | """ wget https://portal.nersc.gov/CheckV/checkv-db-v1.0.tar.gz tar -zxvf checkv-db-v1.0.tar.gz rm checkv-db-v1.0.tar.gz """ |
14 15 16 | """ mkdir -p checkv-db-v1.0 """ |
14 15 16 17 | """ checkv end_to_end ${fasta} -d ${database} -t ${task.cpus} ${confidence_set_name} cp ${confidence_set_name}/quality_summary.tsv ${confidence_set_name}_quality_summary.tsv """ |
19 20 21 22 | """ checkv end_to_end ${fasta} -d ${database} -t ${task.cpus} ${confidence_set_name} cp ${confidence_set_name}/quality_summary.tsv ${confidence_set_name}_quality_summary.tsv """ |
25 26 27 28 29 | """ mkdir negative_result_${confidence_set_name}.tsv echo "contig_id contig_length genome_copies gene_count viral_genes host_genes checkv_quality miuvig_quality completeness completeness_method contamination provirus termini warnings" > ${confidence_set_name}_quality_summary.tsv echo "pos_phage_0 146647 1.0 243 141 1 High-quality High-quality 97.03 AAI-based 0.0 No" >> ${confidence_set_name}_quality_summary.tsv """ |
14 15 16 17 18 19 20 21 22 23 24 25 26 27 | """ # combine if [[ ${set_name} == "all" ]]; then cat *.tsv | grep -v Abs_Evalue_exp | sort | uniq > ${id}.tsv cat *.fasta > ${id}.fasta else cp ${annotation_table} ${id}.tsv cp ${assembly} ${id}.fasta fi # now we remove contigs shorter 1500 kb and very long ones because ChromoMap has an error when plotting to distinct lenghts # we also split into multiple files when we have many contigs, chunk size default 30 filter_for_chromomap.rb ${id}.fasta ${id}.tsv ${id}.contigs.txt ${id}.anno.txt 1500 """ |
44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 | """ #!/usr/bin/env Rscript library(chromoMap) library(ggplot2) library(plotly) contigs <- list() annos <- list() contigs <- dir(pattern = "*.contigs.txt") annos <- dir(pattern = "*.anno.txt") for (k in 1:length(contigs)){ c = contigs[k] a = annos[k] # check if a file is empty if (file.info(c)\$size == 0 || file.info(a)\$size == 0) { next } # check how many categories we have categories <- c("limegreen", "orange","grey") df <- read.table(a, sep = "\\t") set <- unique(df\$V5) if ( length(set) == 2 ) { if ( set[1] == 'High confidence' && set[2] == 'Low confidence') { categories <- c("limegreen", "orange") } if ( set[1] == 'High confidence' && set[2] == 'No hit') { categories <- c("limegreen", "grey") } if ( set[1] == 'Low confidence' && set[2] == 'No hit') { categories <- c("orange", "grey") } } if ( length(set) == 1 ) { if ( set[1] == 'High confidence') { categories <- c("limegreen") } if ( set[1] == 'Low confidence') { categories <- c("orange") } if ( set[1] == 'No hit') { categories <- c("grey") } } p <- chromoMap(c, a, data_based_color_map = T, data_type = "categorical", data_colors = list(categories), legend = T, lg_y = 400, lg_x = 100, segment_annotation = T, left_margin = 100, canvas_width = 1000, chr_length = 8, ch_gap = 6) htmlwidgets::saveWidget(as_widget(p), paste("${id}.chromomap-", k, ".html", sep='')) } """ |
9 10 11 | """ fastp -i ${reads[0]} -I ${reads[1]} --thread ${task.cpus} -o ${name}.R1.fastp.fastq.gz -O ${name}.R2.fastp.fastq.gz """ |
8 9 10 11 | """ mkdir fastqc fastqc -t ${task.cpus} -o fastqc *.fastq.gz """ |
12 13 14 | """ hmmscan_format_table.py -t ${hmmer_tbl} -o ${set_name}_modified """ |
13 14 15 16 17 18 19 20 21 22 23 24 25 | """ if [[ ${params.db} == "viphogs" ]]; then if [[ ${params.version} == "v1" ]]; then hmmscan --cpu ${task.cpus} --noali -E "0.001" --domtblout ${set_name}_${params.db}_hmmscan.tbl ${db}/${db}.hmm ${faa} else hmmscan --cpu ${task.cpus} --noali --cut_ga --domtblout ${set_name}_${params.db}_hmmscan_cutga.tbl ${db}/${db}.hmm ${faa} #filter evalue for models that dont have any GA cutoff awk '{if(\$1 ~ /^#/){print \$0}else{if(\$7<0.001){print \$0}}}' ${set_name}_${params.db}_hmmscan_cutga.tbl > ${set_name}_${params.db}_hmmscan.tbl fi else hmmscan --cpu ${task.cpus} --noali -E "0.001" --domtblout ${set_name}_${params.db}_hmmscan.tbl ${db}/${db}.hmm ${faa} fi """ |
13 14 15 | """ wget -nH ftp://ftp.ebi.ac.uk/pub/databases/metagenomics/viral-pipeline/IMG_VR_2018-07-01_4.tar.gz && tar zxvf IMG_VR_2018-07-01_4.tar.gz """ |
13 14 15 16 17 18 19 20 21 22 | """ if [[ "${set_name}" == "all" ]]; then grep contig_ID *.tsv | awk 'BEGIN{FS=":"};{print \$2}' | uniq > ${name}.tmp grep -v "contig_ID" *.tsv | awk 'BEGIN{FS=":"};{print \$2}' | uniq >> ${name}.tmp cp ${name}.tmp ${name}.tsv generate_counts_table.py -f ${name}.tsv -o ${name}.krona.tsv else generate_counts_table.py -f ${tbl} -o ${set_name}.krona.tsv fi """ |
34 35 36 37 38 39 40 | """ if [[ ${set_name} == "all" ]]; then ktImportText -o ${name}.krona.html ${krona_file} else ktImportText -o ${set_name}.krona.html ${krona_file} fi """ |
12 13 14 15 | """ filter_contigs_len.py -f ${fasta} -l ${params.length} -o ./ CONTIGS=\$(grep ">" ${name}*filt*.fasta | wc -l) """ |
15 16 17 18 | """ sed -i "s/ /|/" ${fasta} mashmap -q ${fasta} -r ${reference} -t ${task.cpus} -o ${confidence_set_name}_mashmap_hits.tsv --noSplit -s ${params.mashmap_len} """ |
15 16 17 18 | """ echo "Downloading ${params.meta_version} of the metadata" wget ftp://ftp.ebi.ac.uk/pub/databases/metagenomics/viral-pipeline/additional_data_vpHMMs_${params.meta_version}.tsv """ |
9 10 11 | """ multiqc -i ${name} . """ |
14 15 16 17 | """ wget -nH ftp://ftp.ebi.ac.uk/pub/databases/metagenomics/viral-pipeline/2022-11-01_ete3_ncbi_tax.sqlite.gz && gunzip -f 2022-11-01_ete3_ncbi_tax.sqlite.gz cp 2022-11-01_ete3_ncbi_tax.sqlite ete3_ncbi_tax.sqlite """ |
18 19 20 21 | """ touch virsorter_metadata.tsv parse_viral_pred.py -a ${fasta} -f ${virfinder} -p ${pprmeta} -s ${virsorter}/Predicted_viral_sequences/*.fasta &> ${name}_virus_predictions.log """ |
14 15 16 17 18 19 20 21 22 23 24 25 26 | """ # get only contig IDs that have at least one annotation hit cat ${tab} | sed 's/|/VIRIFY/g' > virify.tmp IDS=\$(awk 'BEGIN{FS="\\t"};{if(\$6!="No hit"){print \$1}}' virify.tmp | sort | uniq | grep -v Contig) head -1 ${tab} > ${set_name}_prot_ann_table_filtered.tsv for ID in \$IDS; do awk -v id="\$ID" '{if(id==\$1){print \$0}}' virify.tmp >> ${set_name}_prot_ann_table_filtered.tsv done sed -i 's/VIRIFY/|/g' ${set_name}_prot_ann_table_filtered.tsv mkdir -p ${set_name}_mapping_results cp ${set_name}_prot_ann_table_filtered.tsv ${set_name}_mapping_results/ make_viral_contig_map.R -o ${set_name}_mapping_results -t ${set_name}_prot_ann_table_filtered.tsv """ |
16 17 18 19 | """ [ -d "pprmeta" ] && cp pprmeta/* . ./PPR_Meta ${fasta} ${name}_pprmeta.csv """ |
38 39 40 41 42 | """ git clone https://github.com/mult1fractal/PPR-Meta.git mv PPR-Meta/* . rm -r PPR-Meta """ |
15 16 17 | """ prodigal -p "meta" -a ${confidence_set_name}_prodigal.faa -i ${fasta} """ |
14 15 16 17 | """ wget -nH ftp://ftp.ebi.ac.uk/pub/databases/metagenomics/viral-pipeline/hmmer_databases/pvogs.tar.gz && tar -zxvf pvogs.tar.gz rm pvogs.tar.gz """ |
14 15 16 17 | """ [ -d "models" ] && cp models/* . ratio_evalue_table.py -i ${modified_table} -t ${model_metadata} -o ${set_name}_modified_informative.tsv -e ${params.evalue} """ |
12 13 14 15 16 17 18 19 | """ if [[ ${fasta} =~ \\.gz\$ ]]; then zcat ${fasta} > tmp.fasta else cp ${fasta} tmp.fasta fi rename_fasta.py -i tmp.fasta -m ${name}_map.tsv -o ${name}_renamed.fasta rename """ |
13 14 15 16 | """ BN=\$(basename ${fasta} .fna) rename_fasta.py -i ${fasta} -m ${map} -o \${BN}_original.fasta restore 2> /dev/null """ |
14 15 16 17 | """ wget -nH ftp://ftp.ebi.ac.uk/pub/databases/metagenomics/viral-pipeline/hmmer_databases/rvdb.tar.gz && tar -zxvf rvdb.tar.gz rm rvdb.tar.gz """ |
13 14 15 16 17 18 19 20 | """ krona_table_2_sankey_table.rb ${krona_table} ${set_name}.sankey.tsv # select the top ${params.sankey} hits with highest count because otherwise sankey gets messy sort -k1,1nr ${set_name}.sankey.tsv | head -${params.sankey} > ${set_name}.sankey.filtered.tsv tsv2json.rb ${set_name}.sankey.filtered.tsv ${set_name}.sankey.filtered-${params.sankey}.json """ |
37 38 39 40 41 42 43 44 45 46 47 48 | """ #!/usr/bin/env Rscript library(sankeyD3) library(magrittr) Taxonomy <- jsonlite::fromJSON("${json}") # print to HTML file sankey = sankeyNetwork(Links = Taxonomy\$links, Nodes = Taxonomy\$nodes, Source = "source", Target = "target", Value = "value", NodeID = "name", units = "count", fontSize = 22, nodeWidth = 30, nodeShadow = TRUE, nodePadding = 30, nodeStrokeWidth = 1, nodeCornerRadius = 10, dragY = TRUE, dragX = TRUE, numberFormat = ",.3g", align = "left", orderByPath = TRUE) saveNetwork(sankey, file = '${id}.sankey.html') """ |
9 10 11 12 | ''' spades.py --meta --only-assembler -1 !{reads[0]} -2 !{reads[1]} -t !{task.cpus} -o assembly mv assembly/contigs.fasta !{name}.fasta ''' |
14 15 16 17 | """ wget -nH ftp://ftp.ebi.ac.uk/pub/databases/metagenomics/viral-pipeline/hmmer_databases/vpHMM_database_${params.viphog_version}.tar.gz && tar -zxvf vpHMM_database_${params.viphog_version}.tar.gz rm vpHMM_database_${params.viphog_version}.tar.gz """ |
17 18 19 20 | """ run_virfinder.Rscript ${model} ${fasta} . awk '{print \$1"\\t"\$2"\\t"\$3"\\t"\$4}' ${name}*.tsv > ${name}.txt """ |
36 37 38 | """ wget ftp://ftp.ebi.ac.uk/pub/databases/metagenomics/viral-pipeline/virfinder/VF.modEPV_k8.rda """ |
14 15 16 17 18 | """ wget ftp://ftp.ebi.ac.uk/pub/databases/metagenomics/viral-pipeline/virsorter-data-v2.tar.gz tar -xvzf virsorter-data-v2.tar.gz rm virsorter-data-v2.tar.gz """ |
17 18 19 | """ wrapper_phage_contigs_sorter_iPlant.pl -f ${fasta} --db 2 --wdir virsorter --ncpu ${task.cpus} --data-dir ${database} --virome """ |
21 22 23 | """ wrapper_phage_contigs_sorter_iPlant.pl -f ${fasta} --db 2 --wdir virsorter --ncpu ${task.cpus} --data-dir ${database} """ |
14 15 16 17 | """ wget -nH ftp://ftp.ebi.ac.uk/pub/databases/metagenomics/viral-pipeline/hmmer_databases/vogdb.tar.gz && tar -zxvf vogdb.tar.gz rm vogdb.tar.gz """ |
14 15 16 17 | """ wget -nH ftp://ftp.ebi.ac.uk/pub/databases/metagenomics/viral-pipeline/hmmer_databases/vpf.tar.gz && tar -zxvf vpf.tar.gz rm vpf.tar.gz """ |
17 18 19 20 21 22 23 24 25 26 | """ write_viral_gff.py \ -v ${viphos_annotations.join(' ')} \ -c ${quality_summaries.join(' ')} \ -t ${taxonomies.join(' ')} \ -s ${name} \ -a ${fasta} gt gff3validator ${name}_virify.gff """ |
Support
- Share:
-

-

-

- Future updates
Related Workflows





